🤖 Короткий переказ від ШІ
Кістозні захворювання нирок у дітей класифікуються за генетичними та негенетичними причинами, а ультрасонографія є основним методом їх діагностики. Стаття детально розглядає різні форми кістозних захворювань, їх генетику, клінічні прояви та сонографічні особливості, надаючи практичну інформацію для фахівців.
Аналіз охоплює широкий спектр станів, від аутосомно-домінантного полікістозу нирок (АДПН) до кістозної дисплазії нирок, з акцентом на візуалізаційні характеристики. Для ефективної діагностики та диференціації цих станів рекомендовано сучасне медичне обладнання, таке як УЗД апарати, зокрема GE Voluson E8.
✅ Ключові тези зі статті:
- Діагностика: Ультрасонографія є наріжним каменем візуалізації кістозної патології нирок у дітей.
- Лікування: Лікування залежить від типу захворювання, його тяжкості та наявності ускладнень.
- Переваги: Сонографія дозволяє оцінити розмір нирок, диференціацію, характеристики кіст та супутні аномалії, що важливо для діагностики.
- Обмеження: Ранні стадії деяких захворювань можуть мати нормальну сонографічну картину.
Останнє оновлення: 22 липня 2025 р.
Експертна перевірка: Матеріал перевірено та відредаговано експертами RH.ua
Примітка про ШІ: Цей блок створено за допомогою генеративного ШІ для швидкого ознайомлення з основними ідеями статті. Для повного розуміння теми рекомендуємо прочитати повний текст.
⚠️ Не замінює медичну консультацію
 Класифікація кістозних захворювань нирок у дітей заснована на відмінностях між генетичними і негенетичними причинами. Ультрасонограма є наріжним каменем візуалізації кістозної патології нирок.
Класифікація кістозних захворювань нирок у дітей заснована на відмінностях між генетичними і негенетичними причинами. Ультрасонограма є наріжним каменем візуалізації кістозної патології нирок.
АВТОРИ: Laurent Garel, MD
Кістозні захворювання нирок включають в себе численні вроджені чи набуті стани, які призводять до розвитку кіст (порожнин, які покриті епітелієм, заповнені рідиною або напіврідкими речовинами) в одній або обох нирках. Кісти нирки, які відрізняються по етіології, можуть мати морфологічно подібні прояви, тоді як різноманітний спектр аномалій може бути візуалізовано у різних пацієнтів з однаковим етіологічним походженням (наприклад, як при аутосомно-рецесивному полікистозі нирок [АРПН]).
Існує кілька класифікацій кіст нирок з урахуванням патологічних, клінічних і генетичних особливостей. Класифікації, в більшості своїй, необхідні з точки зору більш поглибленого розуміння основ патогенезу походження кіст. Останнім часом було виявлено, що вії трубчастих епітеліальних клітин відіграють важливе значення в сталості диференціації епітеліальних клітин. Відповідно, дефекти первинних апікальних війок трубчастого епітелію, можуть відігравати ключову роль у розвитку кіст.
КЛАСИФІКАЦІЯ КІСТОЗНИХ ЗАХВОРЮВАНЬ НИРОК
З практичної точки зору, клінічно необхідно диференціювати спадкові кістозні ураження нирок від негенетичних кіст нирок (вставка 1).
Вставка 1.
Класифікація кіст нирок
Генетична (спадкова) патологія
– Полікістоз нирок (аутосомно-домінантний, аутосомно-рецесивний)
– Медулярний полікістоз (наприклад, нефронофтіз)
– Гломерулокістозне захворювання нирок (аутосомно-домінантне, спорадичне)
– Синдроми вад розвитку (наприклад, синдроми Мекеля-Грубера, Барді-Бідля, туберозний склероз)
– Мікрокістозні захворювання нирок (вроджений нефротичний синдром)
Негенетична патологія
• Ниркова кістозна дисплазія (полікістозна диспластична нирка, обструктивна ниркова дисплазія)
• негенетичні, недиспластична кіста
– Проста кіста
– Губчаста нирка
– Кістозні новоутворення нирки (наприклад, мультилокулярна кістозна нефрома)
– ”Набуті” кісти (наприклад, хронічна ниркова недостатність)
– Ниркові кісти, нетрубчастого походження (наприклад, кіста ниркового синуса, чашечно-мискової кісти)
Така подвійна класифікація особливо цінна для сонографіста, який проводить дослідження кіст нирок у плодів, новонароджених, дітей і дорослих. Для збереження послідовності в цій статті, кожна патологія розглядається з точки зору її поширеності; генної мутації; клінічних особливостей; історії хвороби і ультразвукових ознак (розмір нирок, загальна ехогенність, збереження або відсутність кортико-медулярної диференціації; розмір кісти, кількість, розташування, пов’язані з нею особливості).
СПАДКОВІ ЗАХВОРЮВАННЯ
Аутосомно-домінантний полікістоз нирок
Поширеність
Поширеність аутосомно-домінантного полікістозу нирок (АДПН) оцінюється в цифрах між 1 на 400 і 1 на 1000 живонароджених.
АДПН є аутосомно-домінантною спадкової рисою з майже 100% пенетрантністю. Ультразвукове дослідження батьків, у випадку з фетальними і педіатричними випадками АДПН, показало високу спонтанну частоту мутації, з даними, що зустрічаються між 5% і 10%, в залежності від серії. Показник в 10% краще корелює з моїм власним досвідом.
Було визначено два гена: PKD1 на хромосомі 16p (85% випадків) і PKD2 на хромосомі 4д (15% випадків). Ген PKD3 також є можливим місцем розвитку, але він ще не ідентифікований. До сих пір всі випадки АДПН плода були пов’язані з PKD1.
АДПН має велику міжродинну і внутрішньородинну варіабельність. Через варіабельність експресивності і спонтанної мутації, сімейний анамнез відсутній у майже 50% педіатричних хворих при обстеженні.
Патогістологія
Будь-який сегмент нефронів і збірних трубочок може бути залучений в патологічний процес. Спочатку більшість кіст при АДПН виникають з дистальної частини нефрона і збірних канальців. На більш пізніх стадіях захворювання, кісти хаотично визначаються в кірковій і мозковій речовині. Хоча двобічне ураження є характерним, 17% випадків є асинхронними або асиметричними, особливо у дітей. До 90% дорослих з АДПН мають кісти печінки. Від дуже незначної в педіатричній віковій групі, частота кіст печінки збільшується з віком. МРТ поширеність кіст печінки при дослідженні полікістоза нирок склала 58% в групі пацієнтів у віці від 15 до 24 років. Кісти можуть також визначатися в підшлунковій залозі (5% пацієнтів); павутинній оболонці (8%) і сім’яних міхурцях (40%). З нашого досвіду, екстраренальні кісти у дітей з АДПН є надзвичайно рідкісними.
Залучення судин в АДПН включає: внутрішньочерепні аневризми, розшарування грудного відділу аорти розсічення і аневризми коронарних артерій. Рутинний пресимптоматичний скринінг внутрішньочерепних аневризм не показаний при відсутності сімейного анамнезу аневризми або субарахноїдального крововиливу.
Клінічні особливості розвитку захворювання
Більшість дітей з АДПН не мають проявів симптомів хвороби. Гіпертонія присутня в приблизно 50% молодих людей (20-34 років) з АДПН. Больовий синдром є найбільш поширеним симптомом у дорослих пацієнтів; гострий біль може бути викликаний кровотечею, каменем або інфекцією. Кровотеча з кісти розвивається частіше, ніж вважалося раніше. Це було показано при дослідженні кісти за допомогою гіперденсивної КТ або МРТ з посиленням сигналу у більш, ніж 90% дорослих пацієнтів з АДПН. Камені в нирках зустрічаються приблизно в 20% випадків у дорослих. Розвиток ниркової недостатності сильно варіює. Мутований ген (PKD1 проти PKD2), розташування мутації в PKD1, поряд з модифікаторами генів мають істотний вплив на клінічний перебіг хвороби. Консиліум по радіологічним методам візуалізації полікістозних захворювань встановив, що об’єми нирки і кісти є кращими предикторами порушення функції нирок. Раннє розпізнавання АДПН у плодів і немовлят не корелює з більш важкими наслідками.
Сонографічні особливості
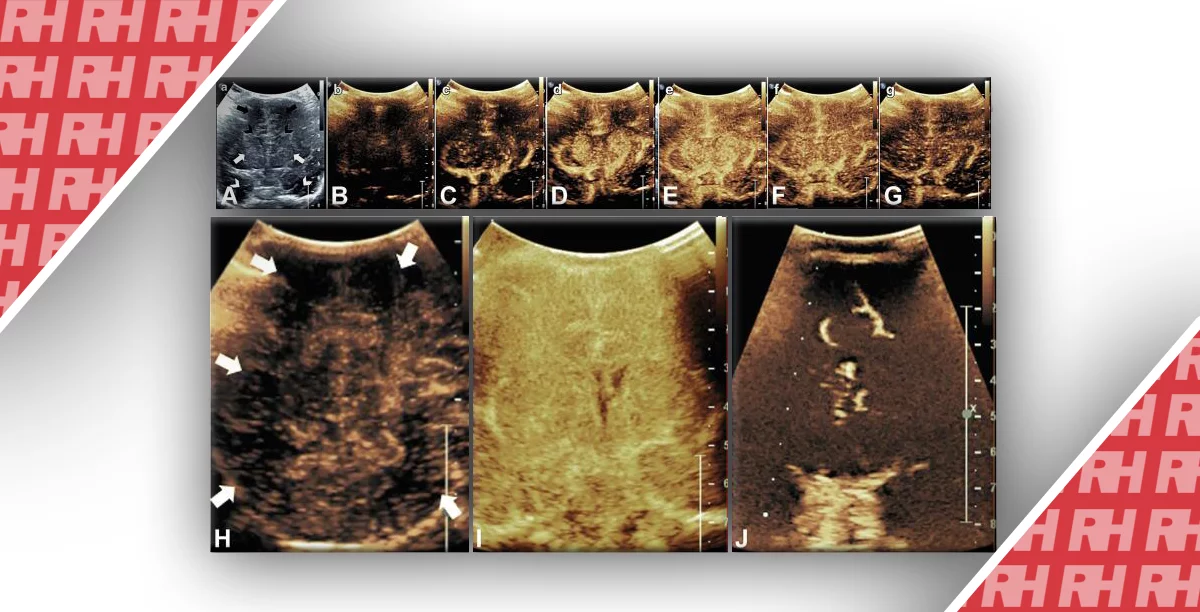
Прояви АДПН у дітей має різноманітну клінічну картину, в основному в залежності від віку пацієнта і генної мутації (Рис. 1-17). У дітей з мутацією PKD1 кісти розвиваються раніше, є більш численними і великими, ніж у дітей з мутацією PKD2. Запропоновано три сонографічних шаблони. (1) Класична форма, яка представлена нирками нормального розміру, з нормально ехогенністю паренхіми і обмеженою кількістю кіст в кірковому або медулярному шарі (див. Рис. 1-5).
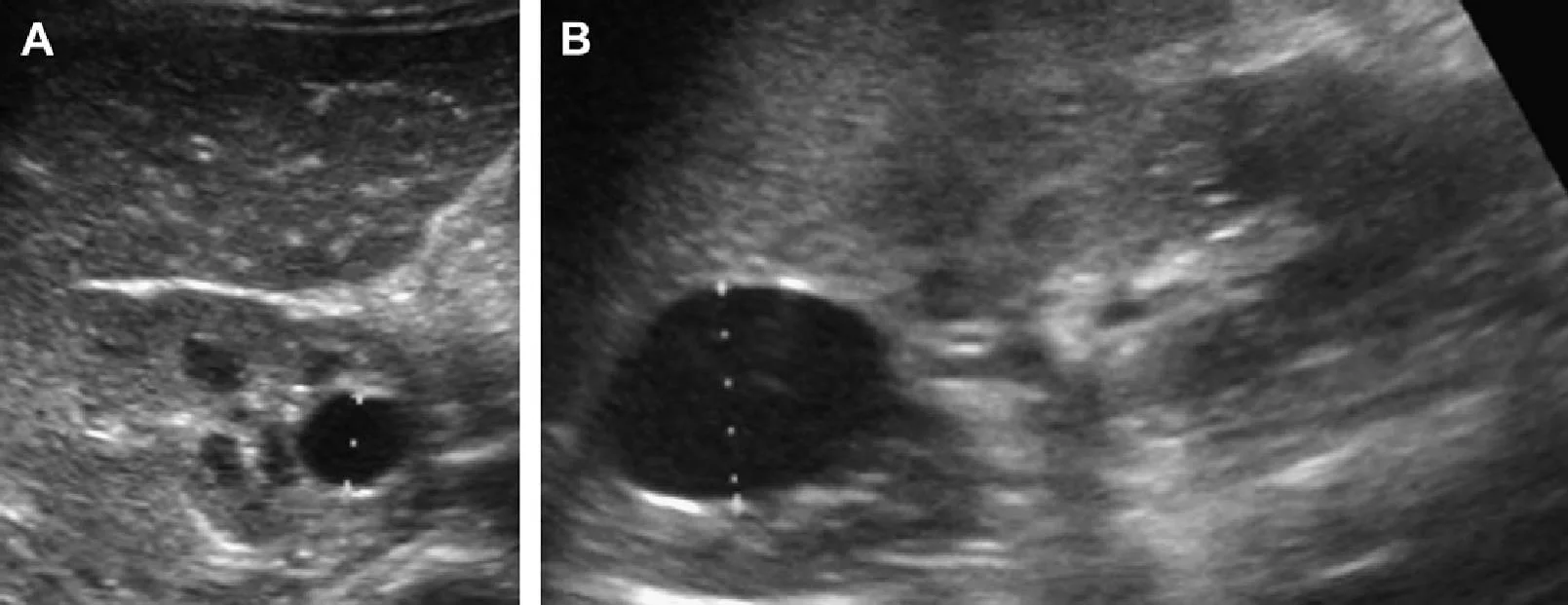
Рис. 1. Трирічний хлопчик обстежується в зв’язку з інфекцією сечовивідних шляхів. Сімейний анамнез з приводу кістозних захворювань нирок відсутній. При сонографії класичні особливості АДПН з двосторонніми кістами (курсори). (А) Права нирка (поперечна проекція). (В) Ліва нирка (поздовжня проекція).
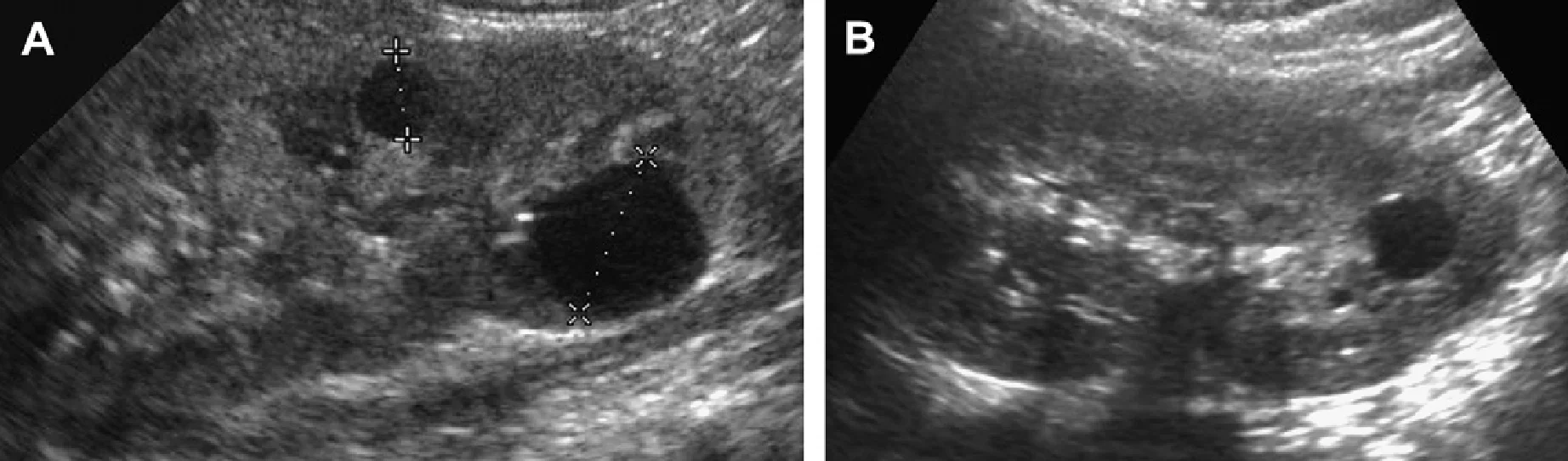
Рис. 2. Мати з відомою патологією АДПН. (A) Поздовжня ультрасонограма її дитини у віці 4 місяців показує дві кісти (курсори) в межах правої нирки. Ліва нирка була нормальною. (В) У віці 3-х років визначається недавно сформована кіста в області нижнього полюса лівої нирки.
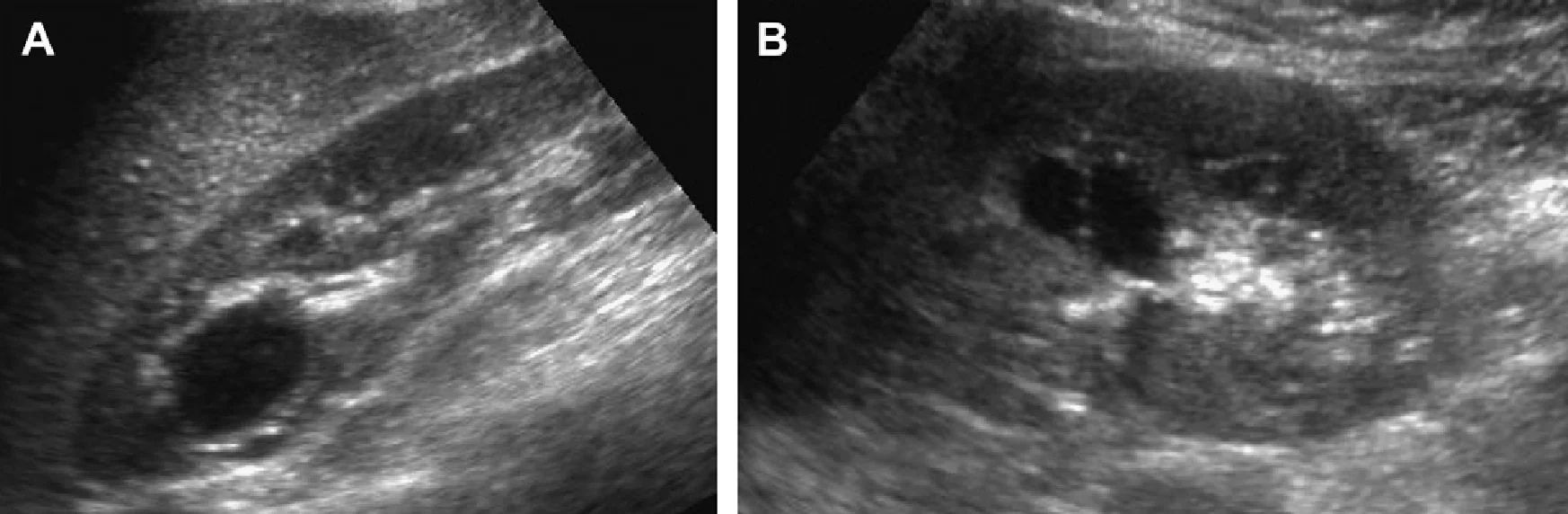
Рис. 3. Восьмирічна дівчинка, чия мати і сестра мають АДПН. Сонограма правої нирки (А) (поздовжня проекція) і ліва нирка (B) (поперечна проекція) показують двосторонні кісти нирок, класичні для АДПН.
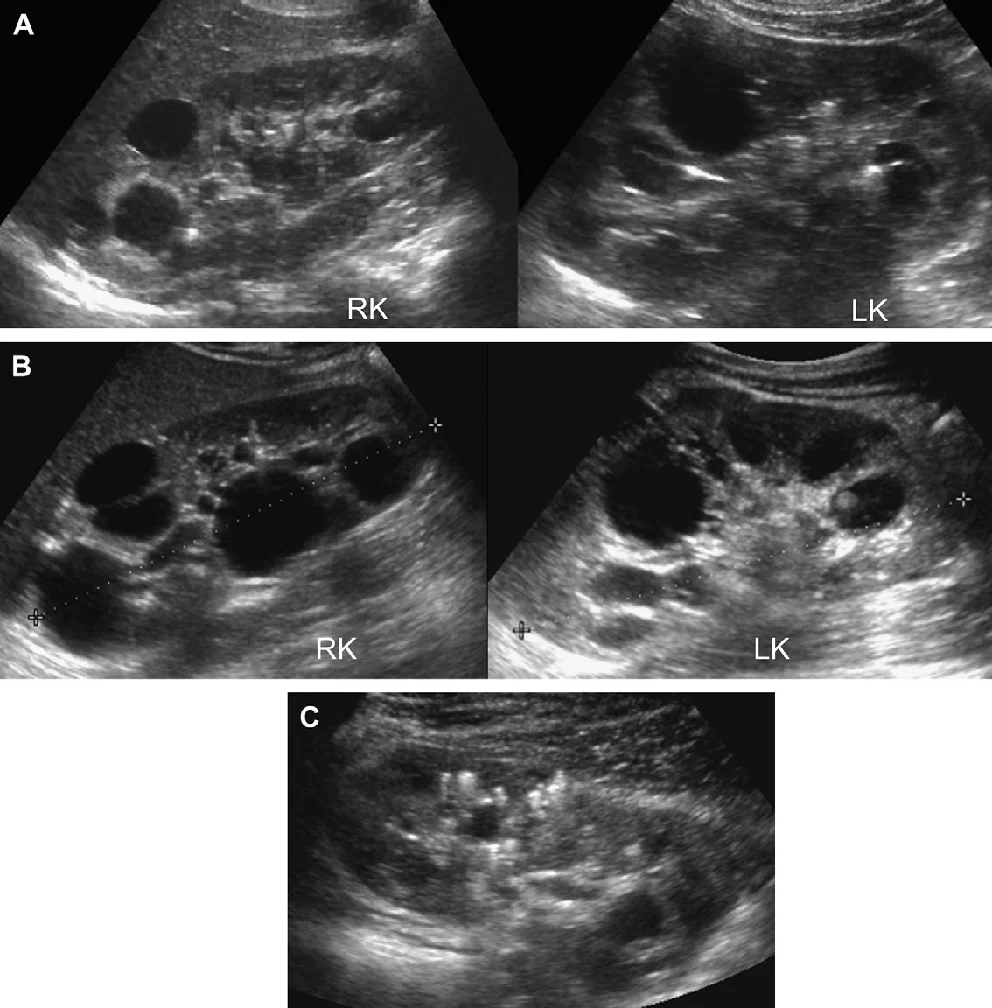
Рис. 4. АДПН з прогресуванням за участю двох експертів. (А) В 9 років, ліва нирка мала розміри 10 см, права нирка 11 см. Кілька білатеральних кіст визначаються в кірковій і мозковій речовині. (В) У 13 років, ліва нирка має розміри 13,1 см, права нирка – 12,7 см. Кісти прогресують в кількості і розмірі. (C) Артефакт «хвіст комети», швидше за все, являє собою відкладення кальцію в кісті.
Проведення сонографії батькам може становити великий інтерес, при відсутності позитивного сімейного анамнезу. Негативні ультразвукові ознаки ураження нирок у обох батьків не виключає АДПН, внаслідок 10% можливості розвитку нової мутації. Спочатку ураження нирок є асиметричним або навіть одностороннім (див. Рис. 6 і 7). Так як кісти можуть проявлятися в будь-якому віці, як правило, не має сенсу піддавати дітей відомих носіїв АДПН рутинному ультрасонографічному обстеженню. (2) Гломерулокістозна форма АДПН в основному розглядається в плодів і немовлят. При цьому нирки значно дифузно збільшені, гіперехогенні і мають ознаки окремих субкапсулярних кіст (див. Рис. 8-10). При відсутності маловоддя, прогноз хороший; в деяких випадках визначається зменшення розміру нирок після народження. (3) При суміжному генному синдромі (PKD1 і TSC2), так як ген PKD1 безпосередньо примикає до гену туберозного склерозу TSC2 на 16-й хромосомі, велика делеція може включати обидві локалізації. За даними сонограми ми маємо як АДПН ознаки, так і туберозного склерозу (див. Рис. 11). При дослідженні мозку часто виявляються позитивні ознаки туберозного склерозу.
Інші ехографічні особливості АДПН можуть зустрічатися в утробі матері і в дитячому віці: помірно збільшені нирки з гіперехогенною кірковою і гіпоехогенною мозковою речовиною (див. Рис 12-15); помірне збільшення нирок без кортико-медулярної диференціації і злегка гіперехогенною паренхімою; також може бути цілком нормальною сонограмою у дітей до десяти років. У моїй практиці, ми рідко виявляли позаниркові кісти у дітей з АДПН (див. Рис. 16 і 17).
Дані літератури, про мінімальну кількість кіст, для того щоб поставити діагноз АДПН в родині з наявністю PKD1, виглядають наступним чином: дві кісти (односторонні або двосторонні) у віці від 15 до 29 років; дві кісти в кожній нирці від 30 до 59 років; чотири кісти в кожній нирці у пацієнтів, яким більше або рівно 60 років.
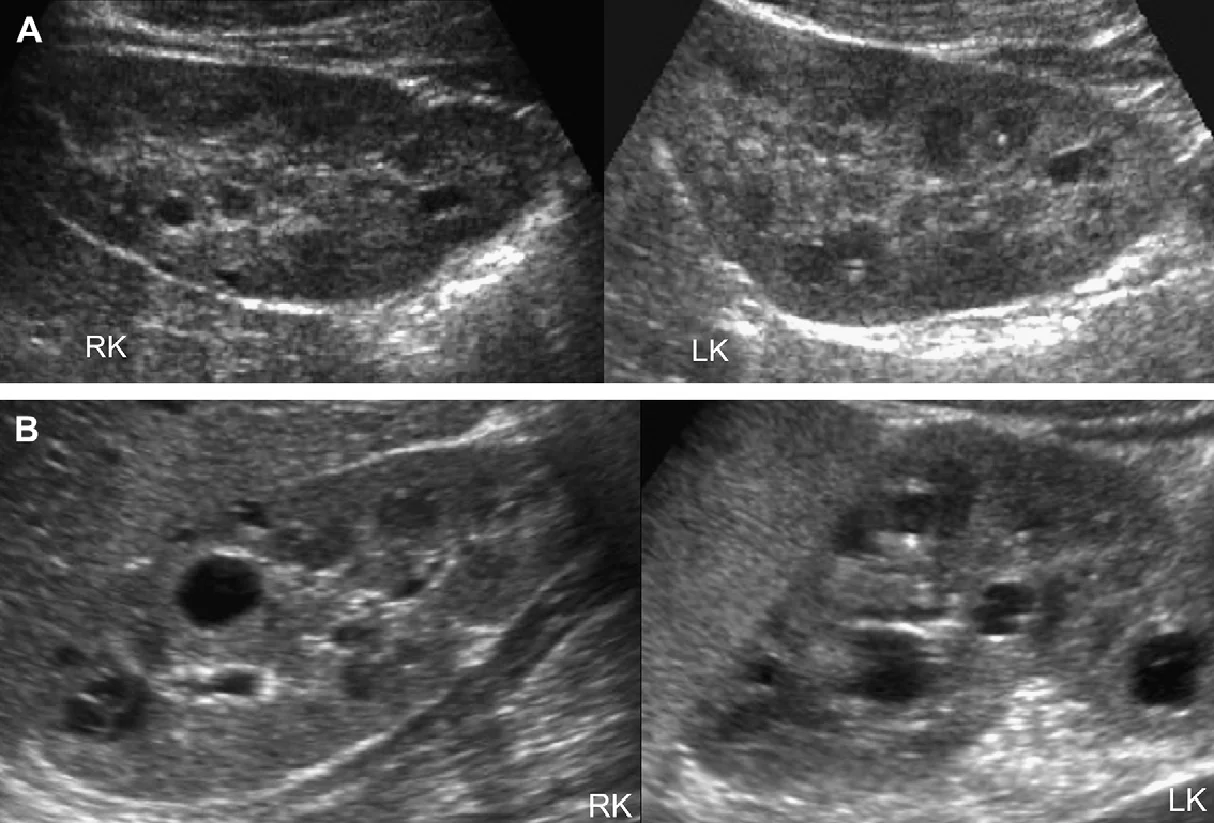
Рис. 5. Ультрасонографія в динаміці у пацієнта, який переніс операцію з приводу везикоуретрального рефлюксу в дитинстві. Ультрасонограма нирок була нормальною до віку 6 років (не показана). Сонограми обох батьків були нормальними, не було ніякого істотного сімейного анамнезу. (А) Поздовжні сонограми обох нирок у дитини 6-річного віку показують невеликі кісти. (В) У 11 років, така АДПН закономірність очевидна в обох нирках.
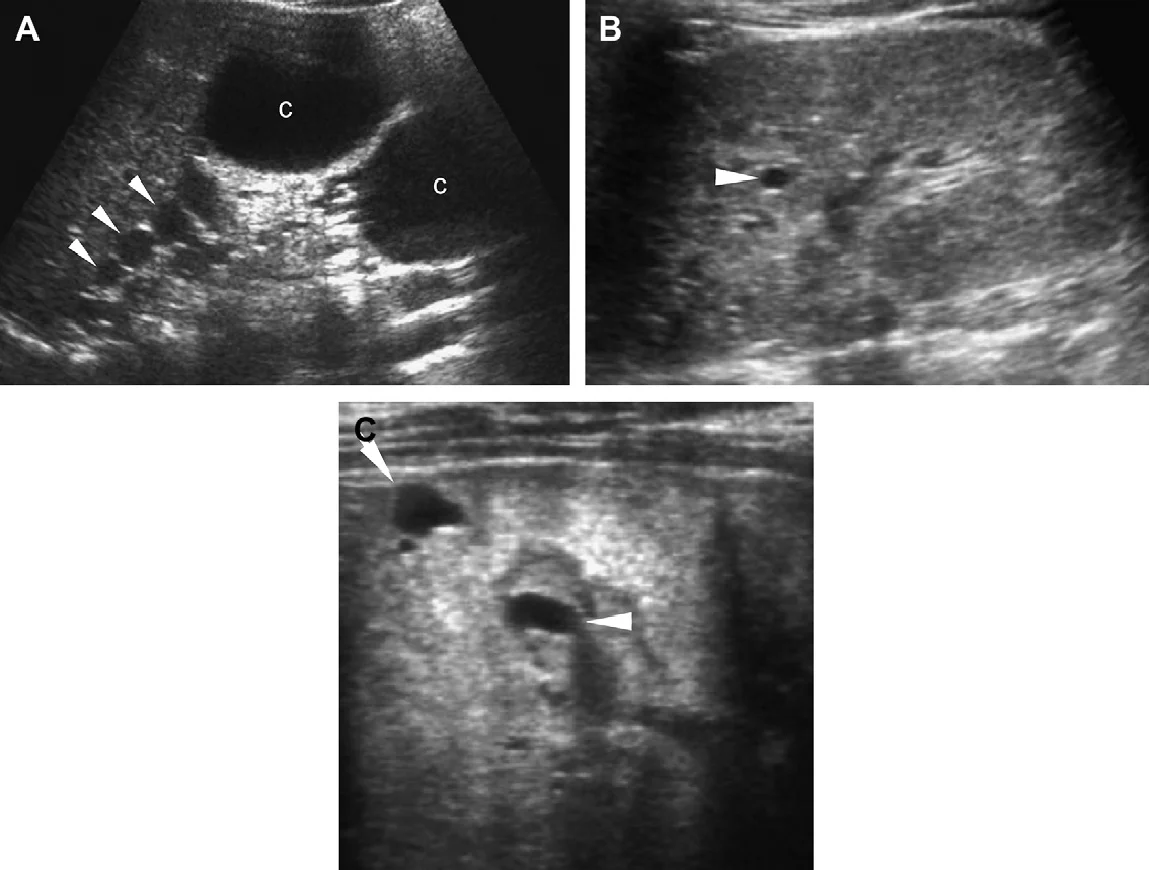
Рис. 6. АДПН з асиметричним ураженням. (А) До 7 років, кісти були видні тільки в правій нирці (дві макрокісти в нижньому полюсі [C], множинні кісти в верхньому полюсі [наконечники стріл]). (B) 7 років після, отримані докази про раннє залучення в кістозний процес лівої нирки (стрілки). (С) Ультрасонограма лівої нирки з високою роздільною здатністю у віці 8 років показує невелику кісту в корі (стрілки) і в мозковій речовині (наконечники стрілки).
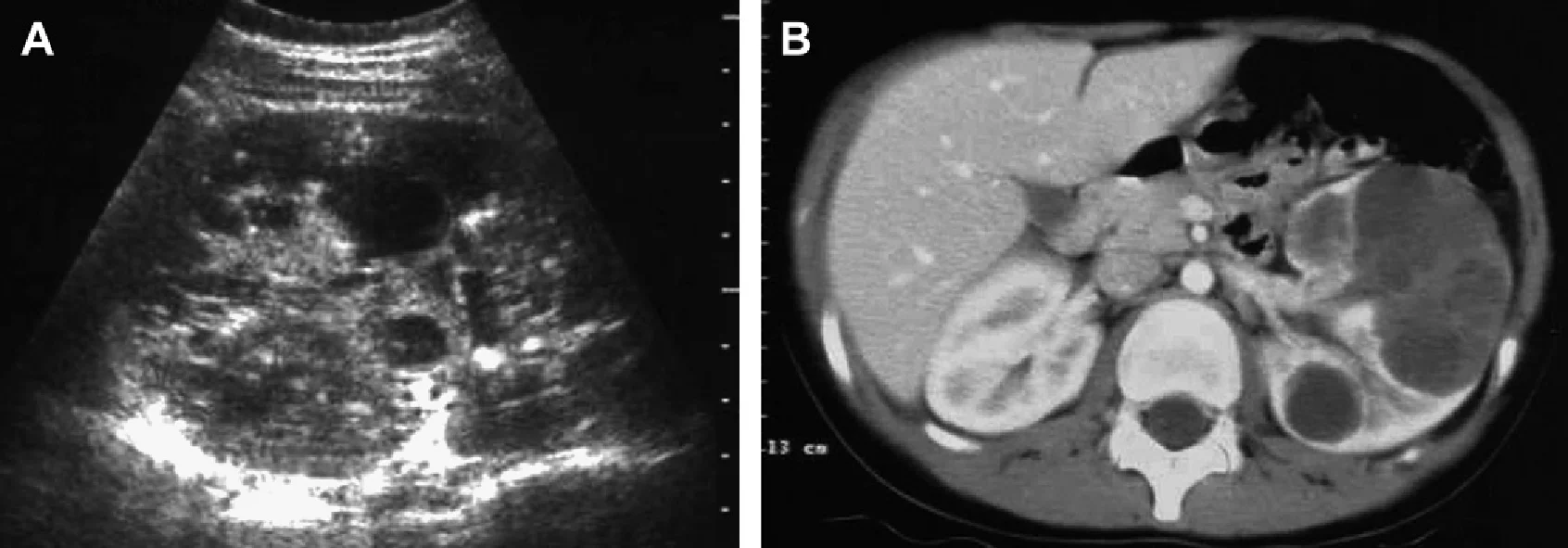
Рис. 7. Односторонній АДПН діагностований у 10-річної дівчинки з посттравматичною гематурією. (А) Ультрасонограма лівої нирки показує множинні кісти і спотворення паренхіми. Права нирка – нормальна. (В) КТ з контрастним посиленням показує велике кістозне утворення в лівій нирці і нормальну праву нирку. Ліва нирка була видалена хірургічним шляхом і гістологічно було доведено одностороннє ураження АДПН. В даному випадку не було ніякого сімейного анамнезу, і ультрасонограми обох батьків були нормальними.
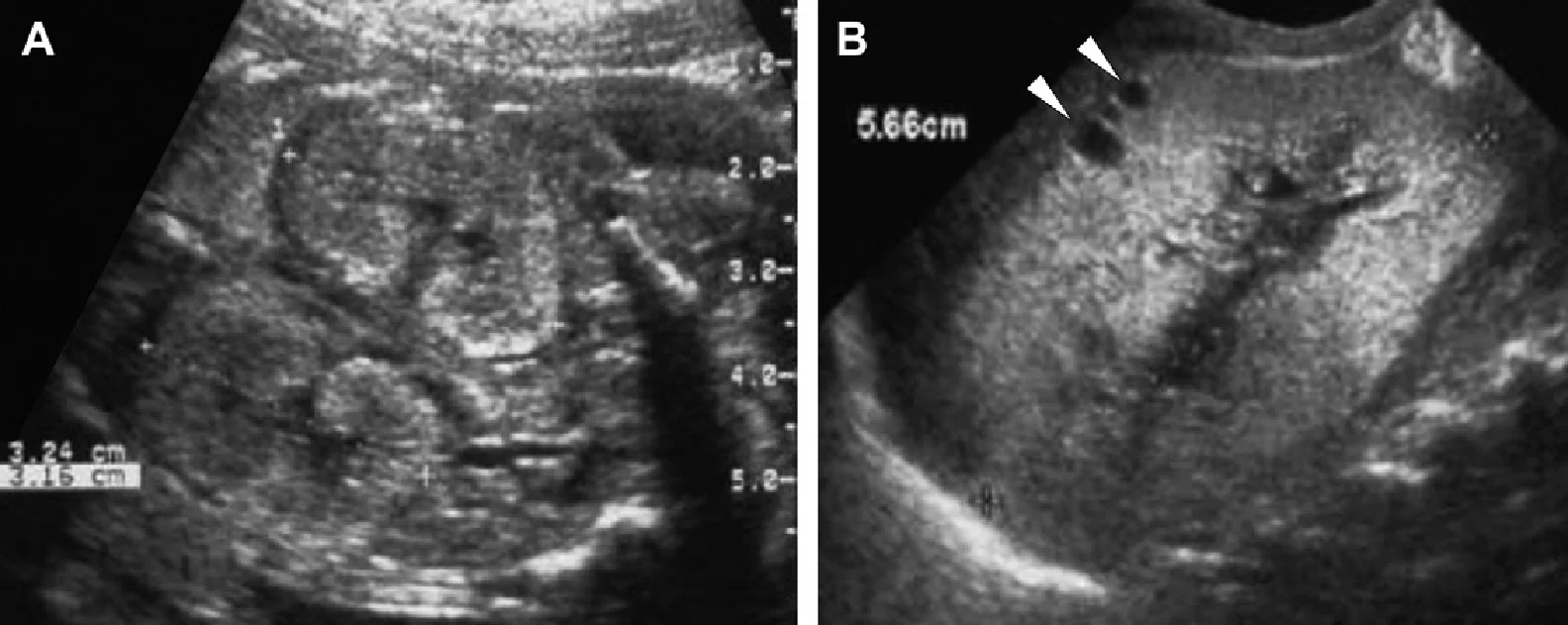
Рис. 8. Гломерулокістозна АДПН форма. (А) Акушерська ультрасонограма на 22 тижні гестації показує невелике збільшення обох фетальних нирок (КУРСОР) з втратою кортико-медулярної диференціації. (В) неонатальна сонограма правої нирки показує дифузно ехогенну нирку з субкапсулярними кістами (наконечники стріл) і відсутність кортико-медулярної диференціації.
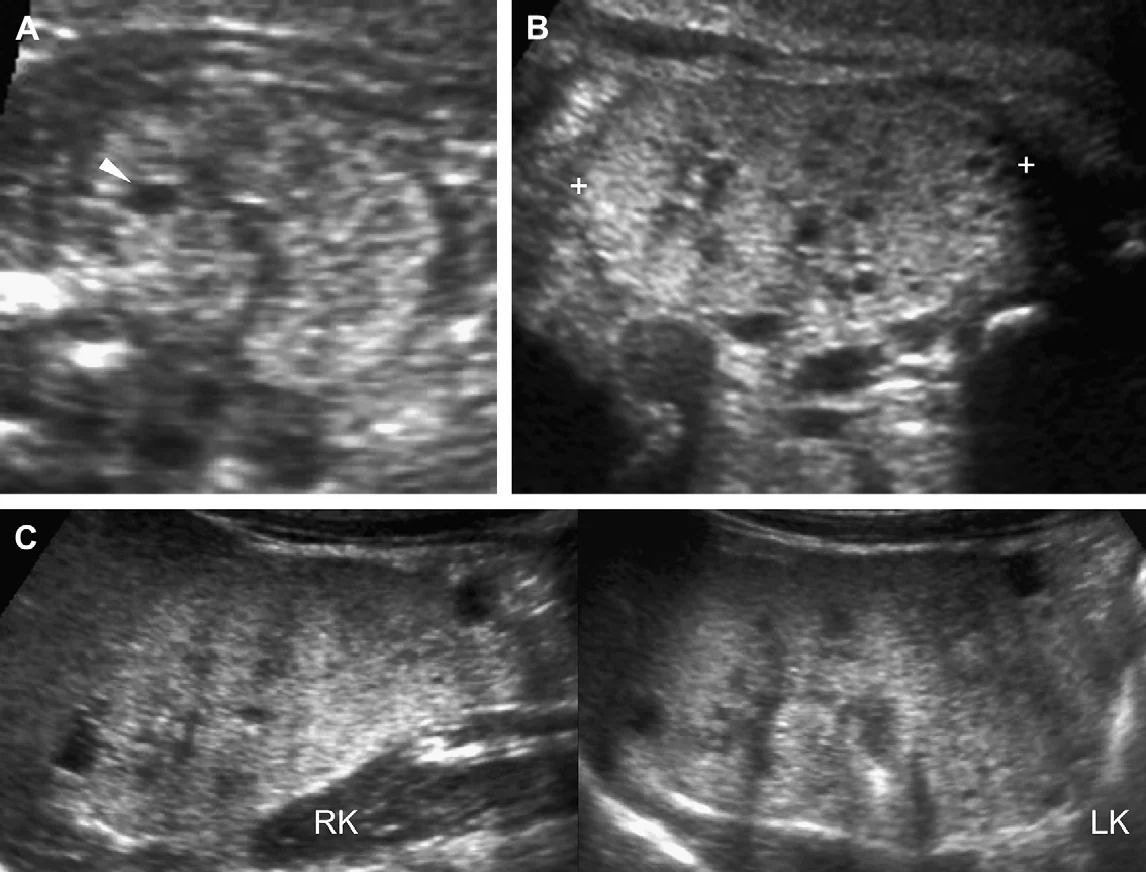
Рис. 9. Гломерулокістозна АДПН форма. (А) Акушерська ультрасонограма на 22 тижні гестації показує нирку з нормальним розміром з однієї кістою (наконечник стріли). (В) Акушерська ультрасонограма у віці 36 тижнів гестації показує помірно збільшену нирку (курсори) (60 мм), відсутність кортико-медулярної диференціації і множинні мікрокісти. (С) Ультразвукове дослідження нирок у віці 5 тижнів показує помірно збільшені гіперехогенні нирки з підкапсулярними кістами.
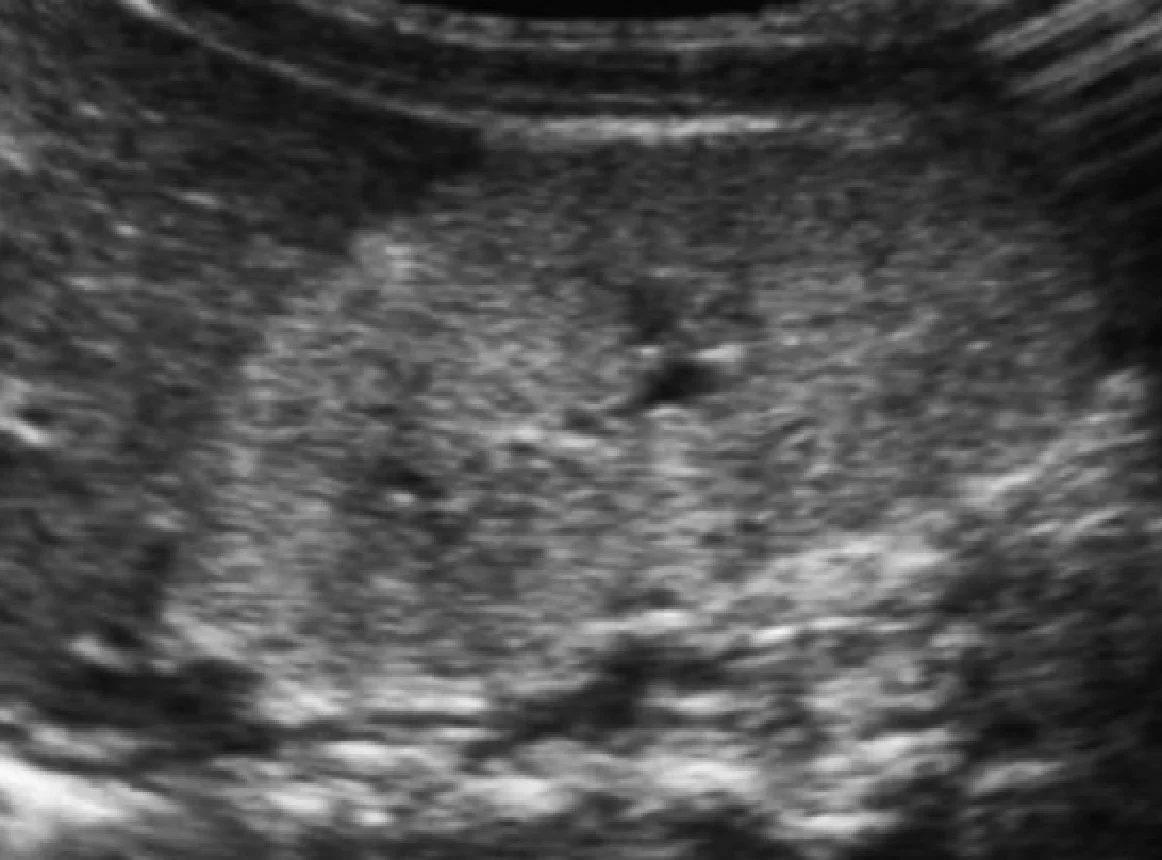
Рис. 10. Гломерулокістозна форма АДПН у новонародженого з позитивним сімейним анамнезом (батько). Ультрасонограма нирок при народженні показує тотально гіперехогенну нирку без видимих субкапсулярних кіст.
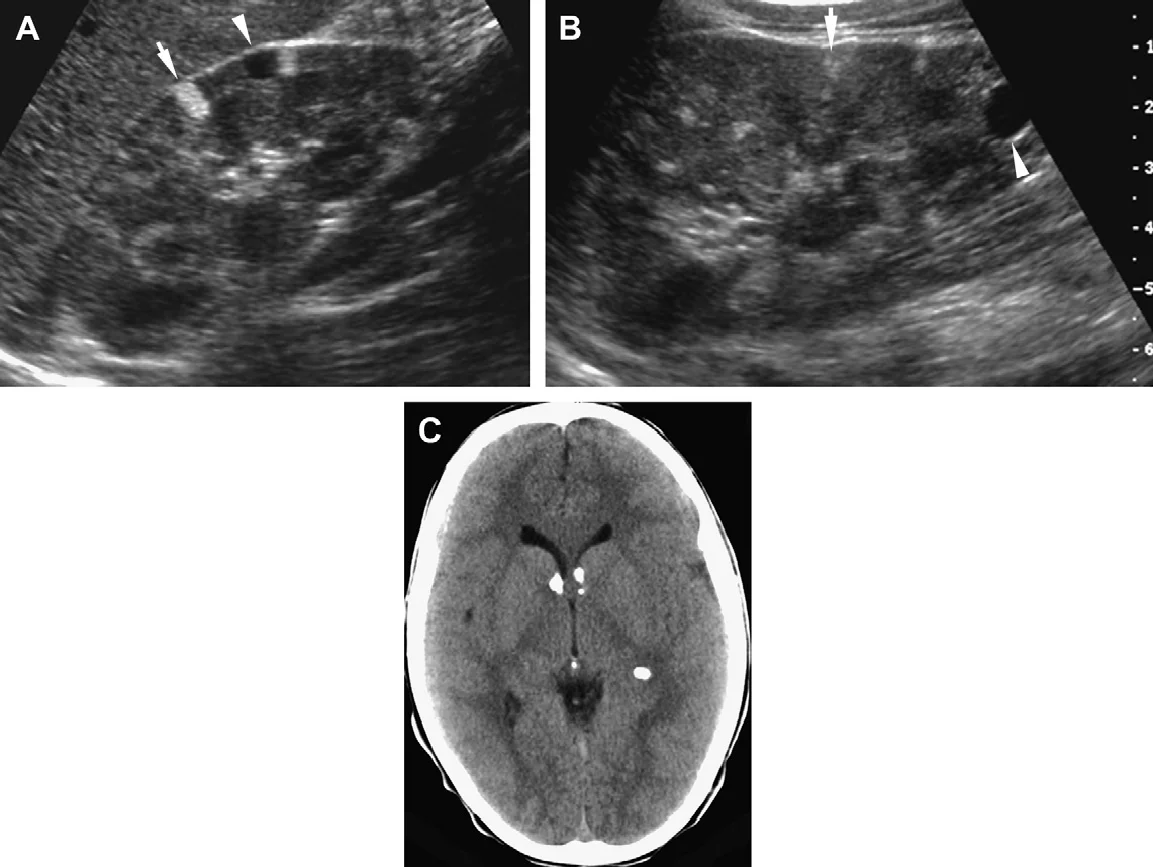
Рис. 11. Туберозний склероз. Восьмирічний хлопчик з ураженнями шкіри. Ультрасонограма нирок показує наявність ангіоліпом (стрілки) і кіст (наконечник стрілки) в корі правої нирки (А) і мозкової речовини в нижній частині лівої нирки (В). (С) КТ головного мозку показує кальциновані субепендимальні вузлики, класичні для туберозного склерозу.
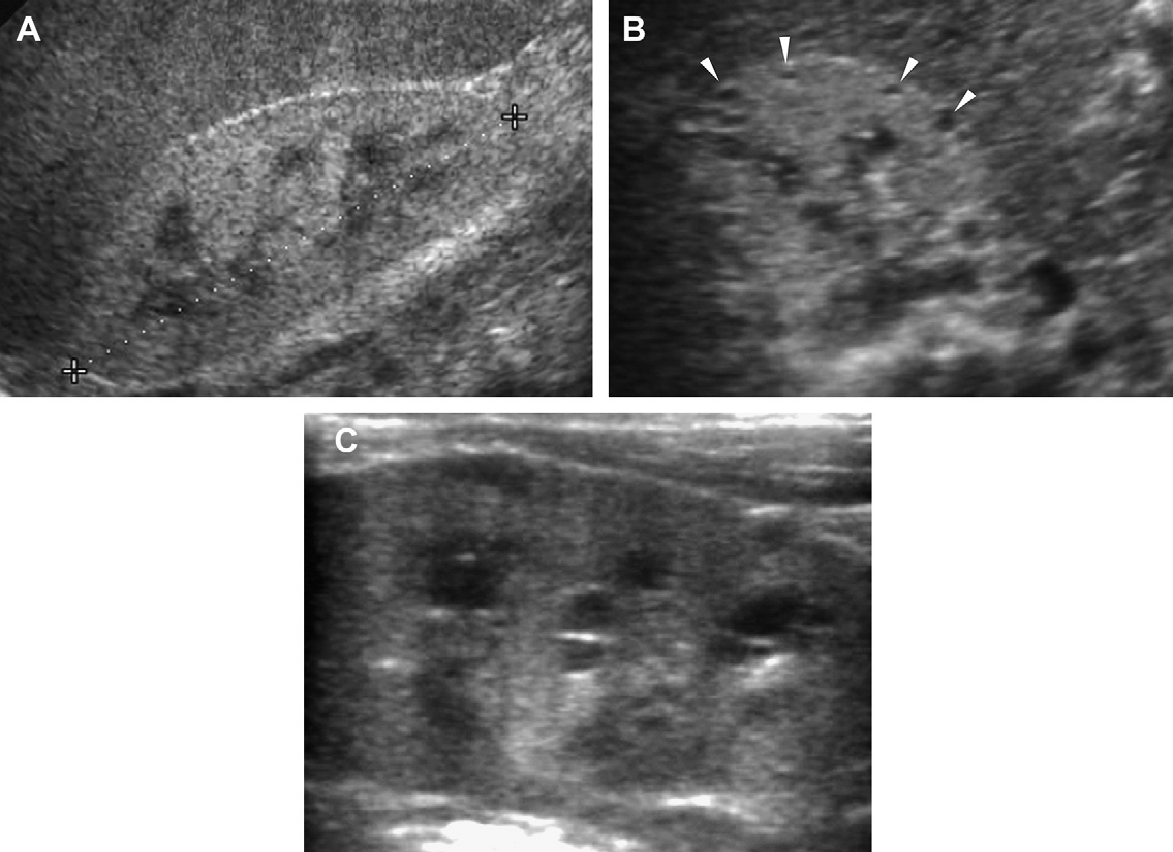
Рис. 12. АДПН. Ниркові порушення були розпізнані на допологовій ультрасонограмі (не відображено). (A) Ультрасонограма нирок на день 1 показує праву нирку (курсори) розміром 6 см; кора – гіперехогенна, але кортико-мозкова диференціація збережена (В). Поперечна сонограма правої нирки через 6 місяців показує численні дрібні кісти (наконечники стрілки). (С) Ультрасонограма з високою роздільною здатністю у віці 1 року демонструє кісти малих розмірів з кортикальною і медулярною локалізацією.
АРПН
Поширеність
Поширеність АРПН за даними більшості літератури знаходиться в межах 1 на 20000 живонароджених.
Генетика
Патологія успадковується за аутосомно-рецесивним типом, АРПН може зустрічатися у рідних братів і сестер (25% потомства батьків-носіїв), але не зустрічається у батьків. АРПН викликається мутаціями в гені PKD1 (ген полікістоза нирок і печінки), який розташований на хромосомі 6p.
PKD1 мутації також є причиною розвитку несиндромального вродженого фіброзу печінки і синдрому Каролі. Як поліцистини при АДПН, білок АРПН – фіброцистин, локалізується в первинної вії ниркових канальців.
Патогістологія
АРПН вражає нирки і печінку, зі специфічним гістологічним проявом обох органів мішеней: кістозна дилатація ниркових канальців; жовчна дисгенезія і фіброз печінки. Як правило, фенотип АРПН в нирці і печінці змінюється зворотно пропорційно. Форма з тяжким ураженням нирок є більш поширеною і проявляється у період новонародженості. І навпаки, форма з більш серйозною печінковою недостатністю і менш важким ураженням нирок зустрічається рідше, і зазвичай проявляється пізніше в дитинстві. При перинатально-неонатальній формі АРПН, нирки значно і симетрично збільшені. Розширення нирок викликано фузіформною дилатацією всіх збірних трубочок в мозковій речовині і в корі.
При більш пізньому прояві АРПН (вроджений фіброз печінки), ураження нирок включає в себе ектазію мозкових проток з мінімальним збільшенням нирок. Дифузні ураження печінки визначаються в портальному просторі: збільшені, фіброзні портальні зони; проліферація і ектазія жовчних проток і гіпоплазія гілок портальних вен.
Іноді, ураження печінки є ізольованим (хвороба Каролі). Хвороба Каролі надзвичайно рідко зустрічається в педіатричній віковій групі.
Клінічні та патогенетичні особливості
При найбільш важкій перинатальній формі, значно збільшені нирки внутрішньоутробно можуть привести до маловоддя, легеневої гіпоплазії або синдрому Поттера. Перинатальне виживання залежить, головним чином, від ступеня легеневої гіпоплазії. Приблизно 30% новонароджених з нирковою предомінантною АРПН вмирають невдовзі після народження. У пацієнтів, які переживають неонатальний період, спостерігається системна артеріальна гіпертензія приблизно в 80% випадків; прогресивне порушення функції нирок призводить до необхідності в трансплантації нирок в різному віці. Як і у дітей з АРПН, ускладнення, пов’язані з ураженням печінки, є більш важливими.
Природжений фіброз печінки може призвести до портальної гіпертензії, а також до біліарного сепсису. Холангіт (лихоманка неясного генезу) може бути первинним проявом у деяких пацієнтів з АРПН. Печінково-клітинна функція рідко порушується. Нирки при переважному ураженні печінки при АРПН можуть бути нормальними або злегка збільшеними, а також можуть мати ознаки ектазій мозкових проток різного ступеня або макрокістозне ураження.
Останні серії досліджень показали, що прогноз АРПН для дітей, які виживають в неонатальному періоді і в перші місяці життя, не такий сумний, як вважається класично.
Сонографічні особливості
Сонографічні моделі АРПН досить варіабельні, але мають характерні особливості в більшості випадків, особливо коли визначаються ознаки ураження одночасно в нирках і печінці (Рис. 18-28). Датчики з високою роздільною здатністю в даній ситуації просто необхідні.
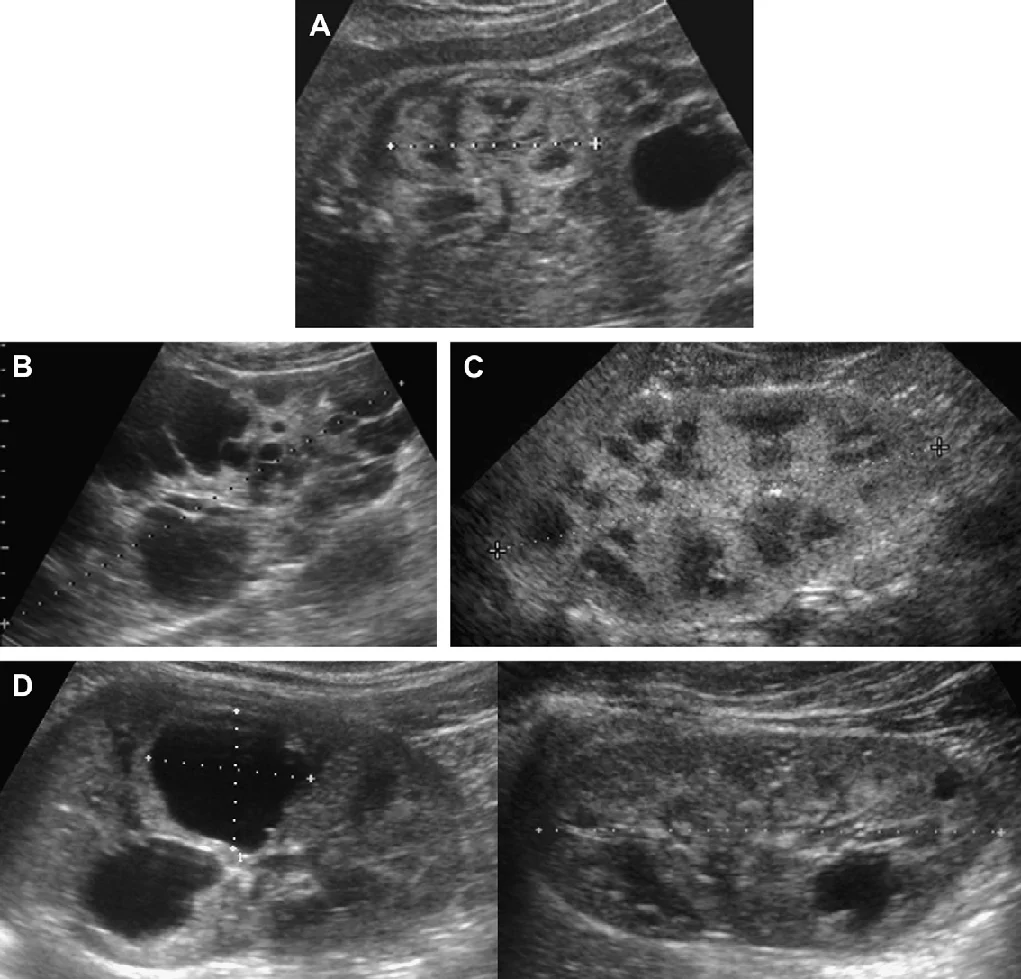
Рис. 13. АДПН. Випадок описує аномальні фетальні нирки. (А) Акушерська ультрасонограма на 34 тижні гестації показує ехогенну кору в нирці зі збереженою кірково-медулярною диференціацією. (В) Ультрасонограма нирок матері виявила раніше невідомий АДПН. (C) Ультрасонограма нирок новонародженого показує ехогенну нирку (курсори), 57мм довжиною з виразним мозковим шаром. (D) Поздовжня ультрасонограма в 3 роки показує характерні риси АДПН, які присутні в обох нирках.
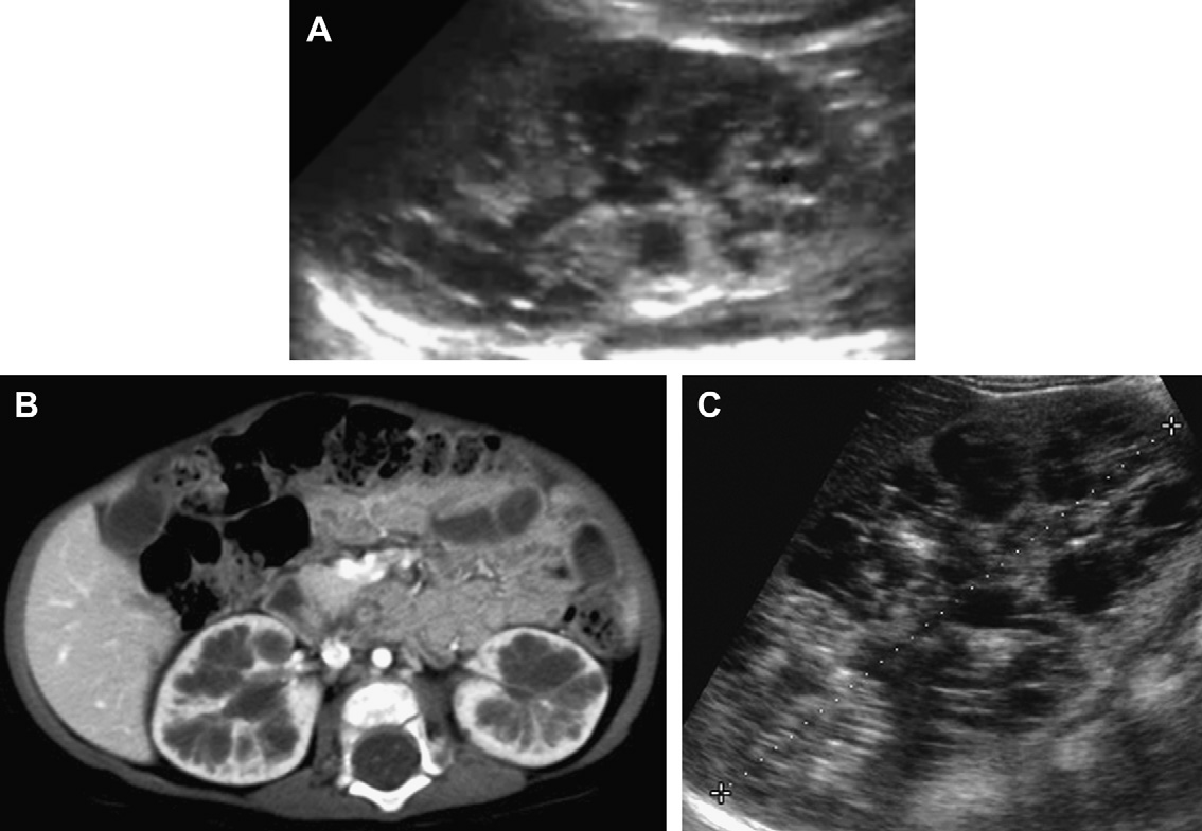
Рис. 14. АДПН з переважанням раннього залучення медулярної зони в патологічний процес. Відповідний сімейний анамнез відсутній. (А) Ультрасонограма, яка виконана в 5-місячному віці в зв’язку з інфекцією сечовивідних шляхів, показує кісти в межах медулярної зони. (В) КТ з контрастним посиленням демонструє ті ж гіподенсивні осередки медулярної зони. Хірургічна біопсія правої нирки довела АДПН. (C) Ультрасонограма при динамічному спостереженні протягом 3-х років показує, що множинні кісти збільшилися в межах пірамід медулярної зони (довжина правої нирки = 11 см).
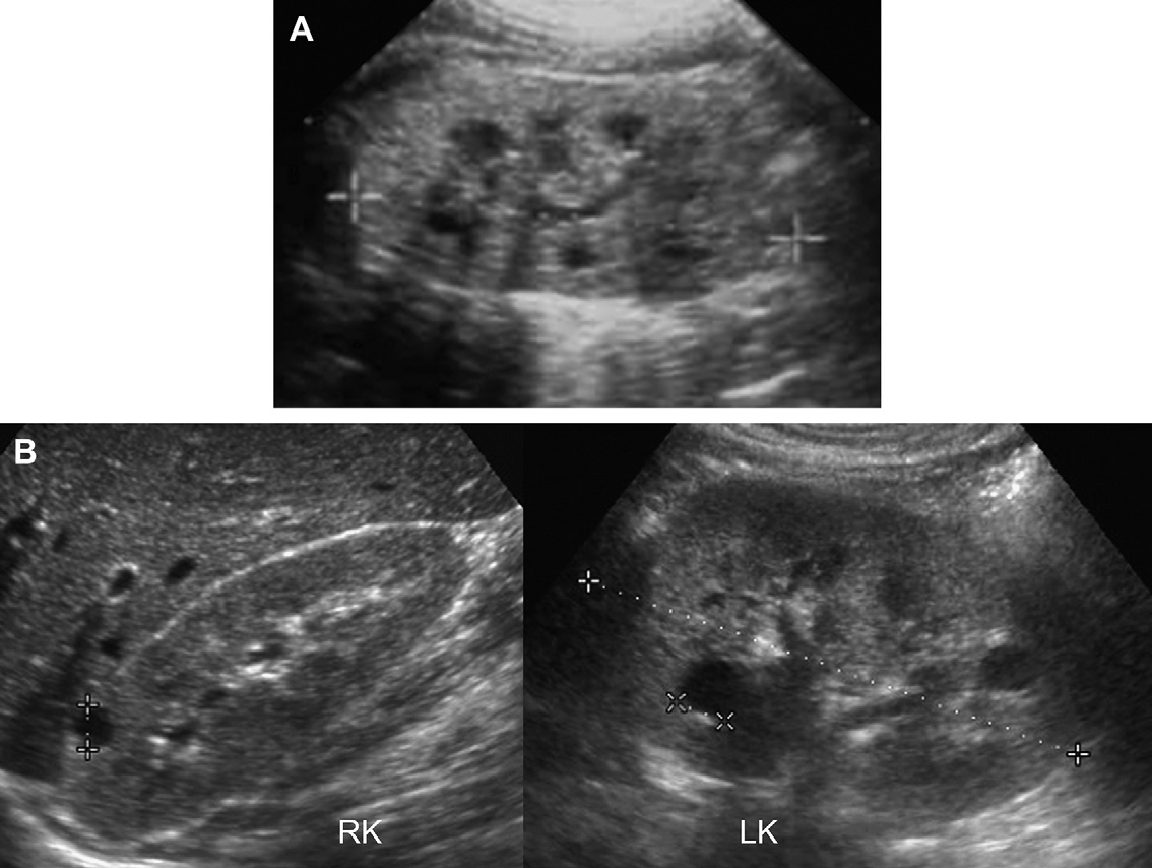
Рис. 15. АДПН з повільним прогресом при послідовному дослідженні. (А) Ультрасонограма новонародженого показує злегка збільшену нирку з ехогенною корою і гіпоехогенною мозковою речовиною. (B) Подальше дослідження в 15-річному віці показує слабкі зміни проявів АДПН в обох нирках.
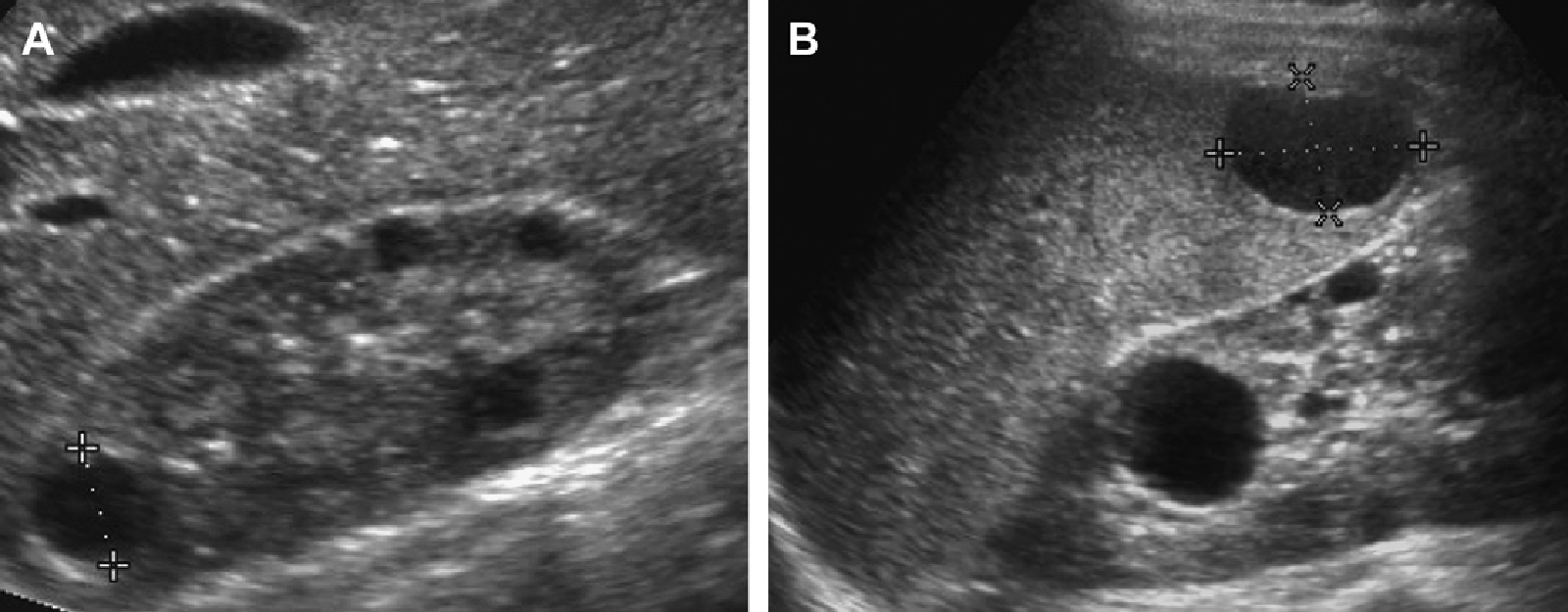
Рис. 16. АДПН 15-річного пацієнта. (А) Ультрасонограма нирок показує кісти в кірковій і мозковій речовині правої нирки. (В) Зображення в лівому верхньому квадранті показує одиночну кісту нижнього полюса селезінки (курсорів), поряд з кістами в лівій нирці.
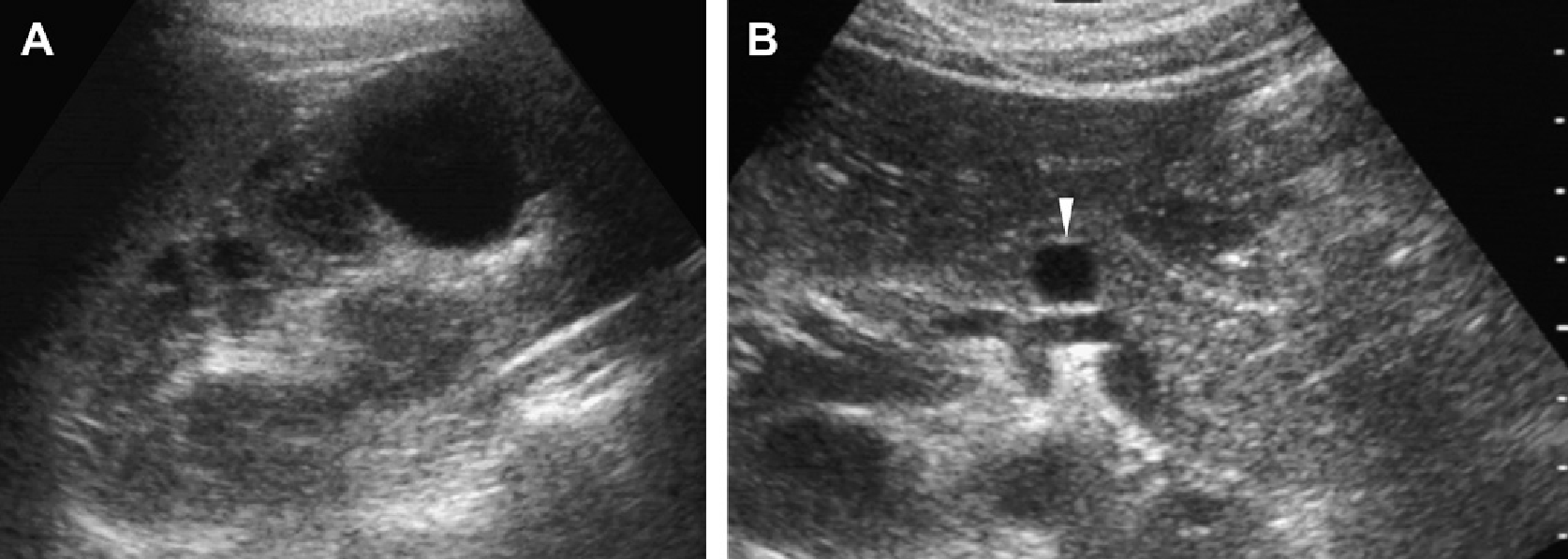
Рис. 17. АДПН 12-річного пацієнта. (A) Поздовжня ультрасонограма лівої нирки показує множинні кісти. (B) Одиночна 8-мм кіста (наконечник стріли) визначається в тканині підшлункової залози.
Особливості нирок при АРПН
Внутрішньоутробно і при народженні спостерігаються: виражена двостороння нефромегалія з дифузною гіперехогенністю кіркової і мозкової речовини, втрата кортико-медулярної диференціації, нечітка візуалізація системи збірних канальців, при збережених контурах нирки. У хворих, які виживають, ультрасонограма показує скорочення ниркової довжини з плином часу. Помірна двостороння нефромегалія з гіпоехогенним зовнішнім кортикальним обідком, збережена кортико-мозкова диференціація, мозкові макрокісти, ехогенні, що не дають тіні, точки або вогнища в медулярній зоні, зазвичай визначаються у дітей і підлітків. Обидві ехографічні моделі нирок при АРПН (величезні яскраві нирки, псевдомедулярний нефрокальциноз в помірно збільшених нирках) високоспецифічні ознаки для постановки діагнозу. Ці дві моделі можуть бути діагностовані в різні періоди життя у одного і того ж хворого.
Особливості печінки при АРПН
Гепатомегалія з переважанням лівої частки, збільшена ехогенність печінки (перипортальне потовщення), жовчні кісти або фокальна дилатація є сонографічними ознаками вродженого фіброзу печінки. Портальна гіпертензія, яка є ускладненням вродженого фіброзу печінки, призводить до спленомегалії і розвитку венозних колатералей.
Інші методи візуалізації
КТ і МРТ можуть бути корисними методами дослідження в окремих випадках при візуалізації нефромегалії, кістозної дилатації ниркових канальців (“покреслена”’ структура, мозкові кісти), гепатоспленомегалії, периферичної біліарної дилатації і кіст. Через ризик розвитку холангіту після процедури, черезшкірне або ретроградне контрастування жовчних проток протипоказано у пацієнтів з АРПН.
НЕФРОНОФТІЗІС
Поширеність
Нефронофтізіс і медулярна кістозна хвороба мають одні і ті ж патологічні і клінічні особливості. Вони відрізняються за способом їх успадкування (аутосомно-рецесивний і аутосомно-домінантний), віком виникнення і кінцевою стадією ниркової недостатності, а також екстраренальними проявами. Нефронофтізіс є досить рідкісним патологічним станом, який реєструється в 1-2 випадках на 100000 живонароджених. На нього припадає від 10% до 20% випадків ниркової недостатності в дитинстві.
Генетика
Нефронофтізіс – аутосомно-рецесивне захворювання, є генетично неоднорідним, при цьому було ідентифіковано вісім причинних генів. NPHP1 генні мутації на хромосомі 2 визначаються у 50% – 80% пацієнтів. Повідомляється про мутації більш ніж одного гена. Передбачається також, що 15% нових мутацій відбуваються в нефронофтізіс – медулярному кістозному комплексі. Гени NPHP нефроцистини кодують локалізуючись в первинних віях ниркових канальців епітеліальних клітин, а також в фоторецепторних віях (що пояснює асоціацію нефронофтізіса з пігментною ретинопатією).
Патогенез
Нефронофтізіс характеризується хронічним дифузним тубулоінтерстіціальним нефритом, який прогресує до фіброзу і термінальної ниркової недостатності. Невеликі кісти в зоні кортико-медулярного співустя або в межах мозкової речовини стають видимими на пізній стадії розвитку патологічного процесу. Ці кісти виникають з дистальних звивистих канальців і збірних трубочок. Трубчасті базальні мембрани, як правило, потовщені, що характерно для ювенільної і підліткової форм нефронофтізіса.
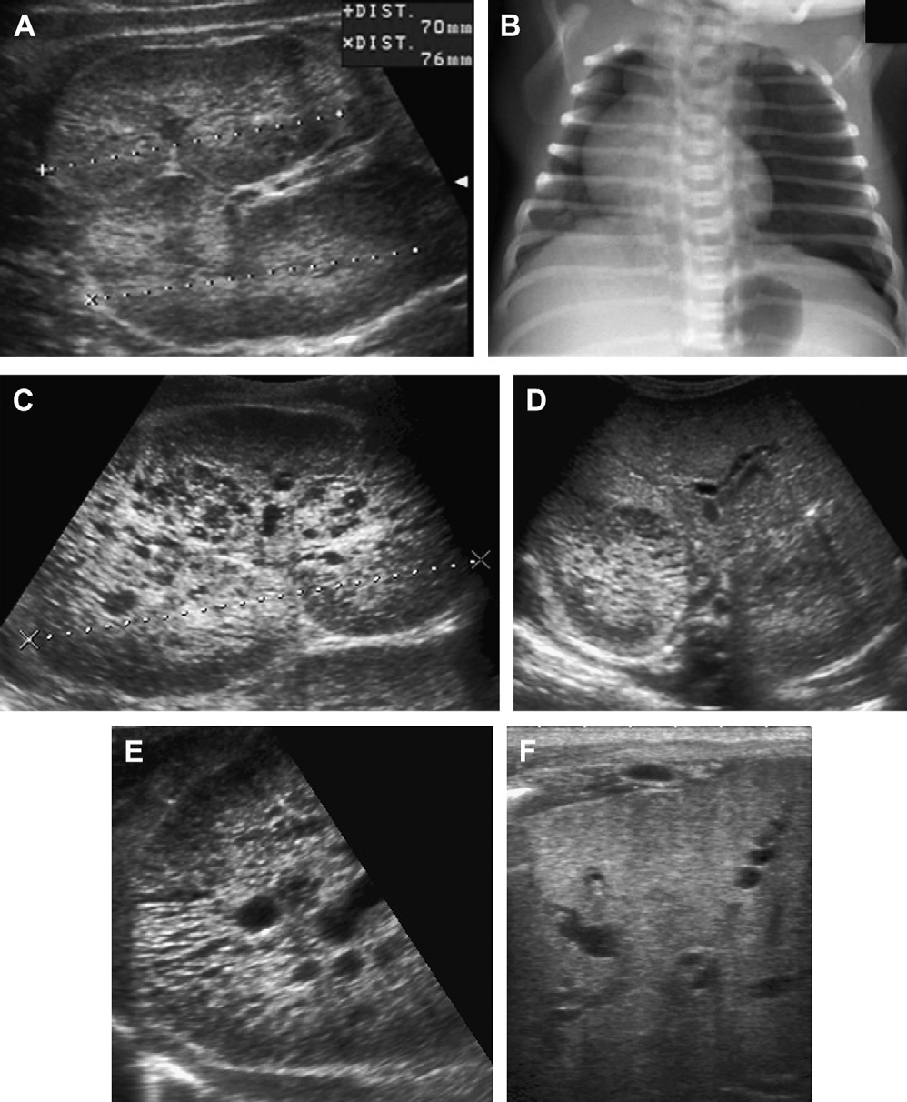
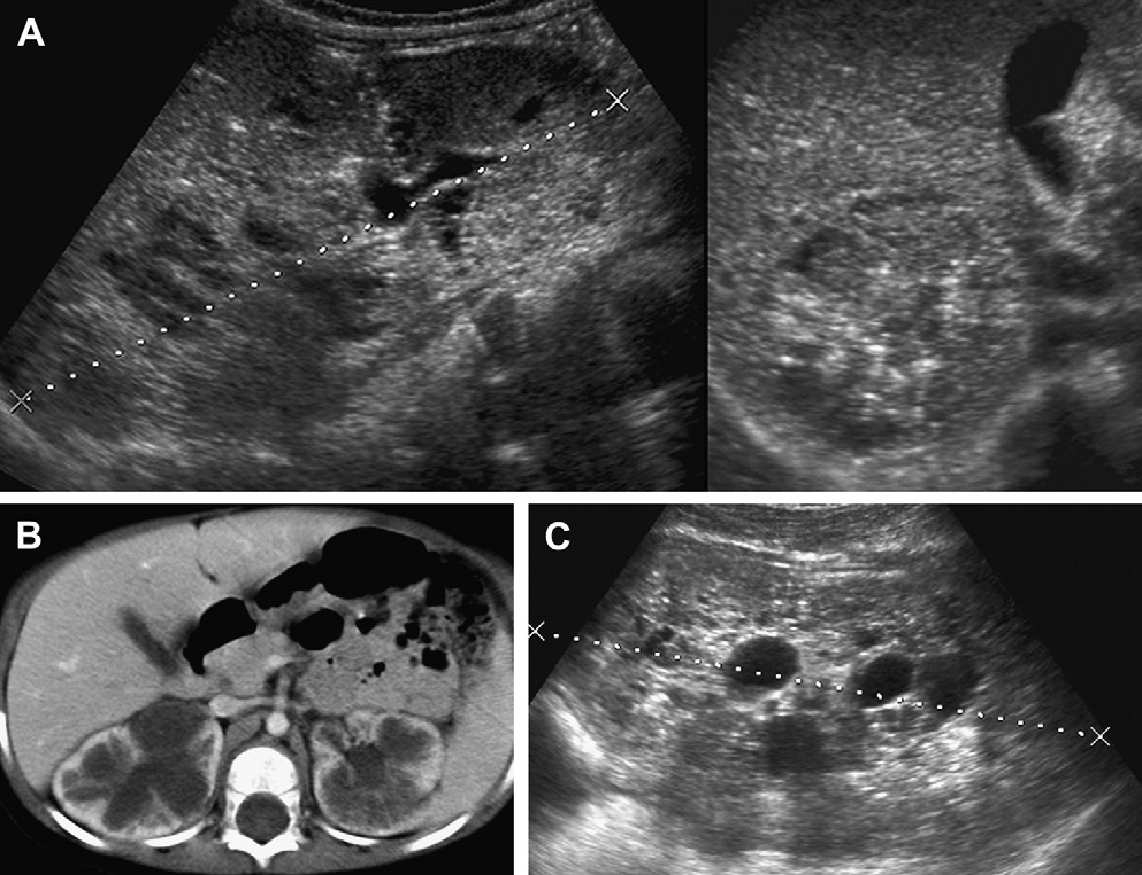
Рис. 18. АРПН, неонатальна форма. (А) Акушерська ультрасонограма на 30 тижні гестації показує ознаки двосторонньої нефромегалії. Відзначено також маловоддя і малі розміри грудної клітини плоду. (B) Рентгенограма при народженні показує двосторонній пневмоторакс і базальну легеневу гіпоплазію. (C) Поздовжня ультрасонограма правої нирки при народженні показує помітно збільшену нирку (до 9 см) з дифузійною трубчастою дилатацією в кірковій і мозковій речовині. (D) Поперечна ультрасонограма показує ехогенну праву нирку і асоційовану біліарну ектазію. (Е) Ультрасонограма нирки з високою роздільною здатністю показує множинні розширені канальці нирок і мозкові кісти. (F) Ультрасонограма печінки з високою роздільною здатністю показує тубулярну ектазію і невеликі паренхіматозні кісти. Незважаючи на агресивну терапію, пацієнт помер у віці 5 тижнів.
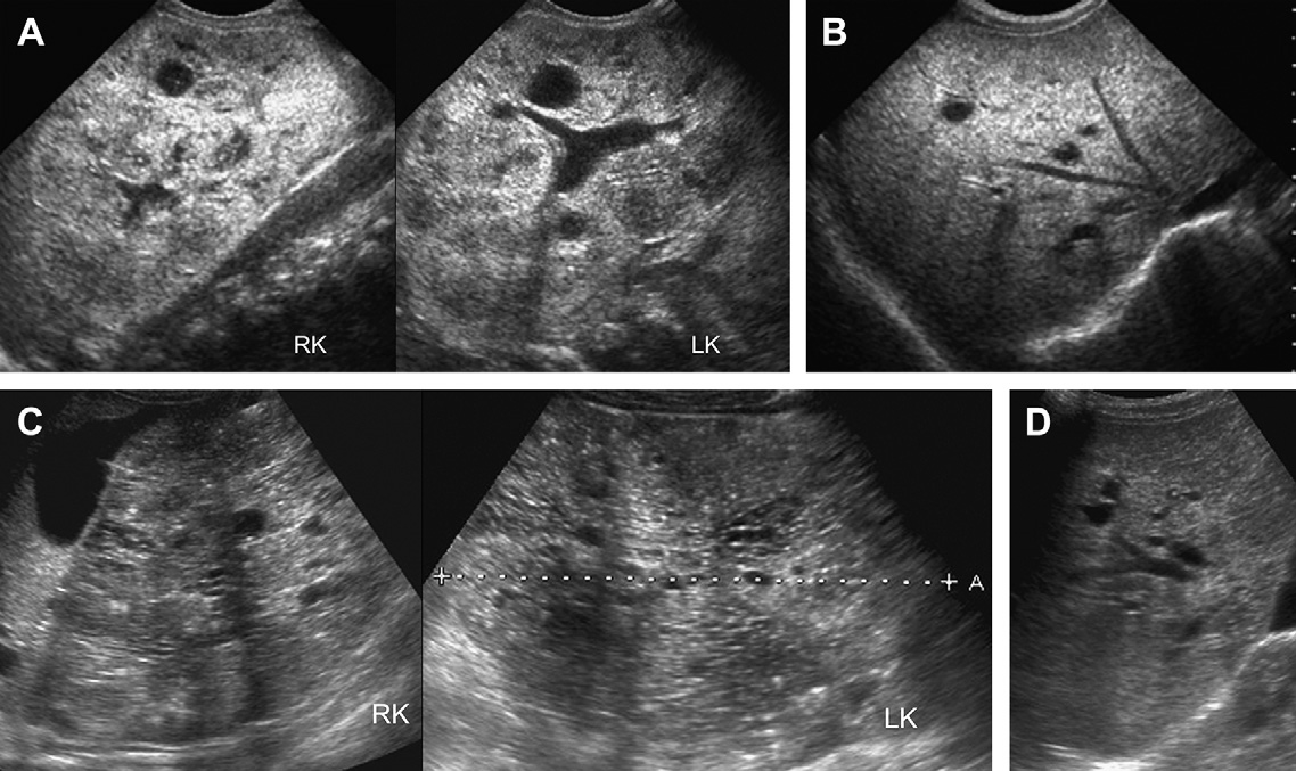
Рис. 19. АРПН, неонатальна форма, ультрасонографія в динаміці. (А) Поздовжня і поперечна ультрасонограма нирок на 2-й день життя показує двосторонню нефромегалію з гіперехогенними і медулярними кістами. (В) Печінка помірної ехогенності і містить невеликі кісти. (C) У 6 років, ультрасонограма показує збільшені нирки (19 см в довжину) з незліченними крихітними кістами і втратою кірково-медулярної диференціації. (D) Асоційована біліарна дисгенезія характерна для АРПН.
Рис. 20. АРПН, неонатальна форма, ультрасонографія в динаміці. (А) Поздовжня і поперечна ультрасонограма правої нирки в 1,5 року показує збільшену нирку (12 см) з медулярною кістозною дилятацією і ехогенними точками. (В) КТ з контрастуванням для порівняння проведена в тому ж віці. (C) Поздовжня ультрасонограма нирок у віці 8 років виявила, що мозкові макрокісти стали більш виразними на тлі 16-сантиметрової нирки.
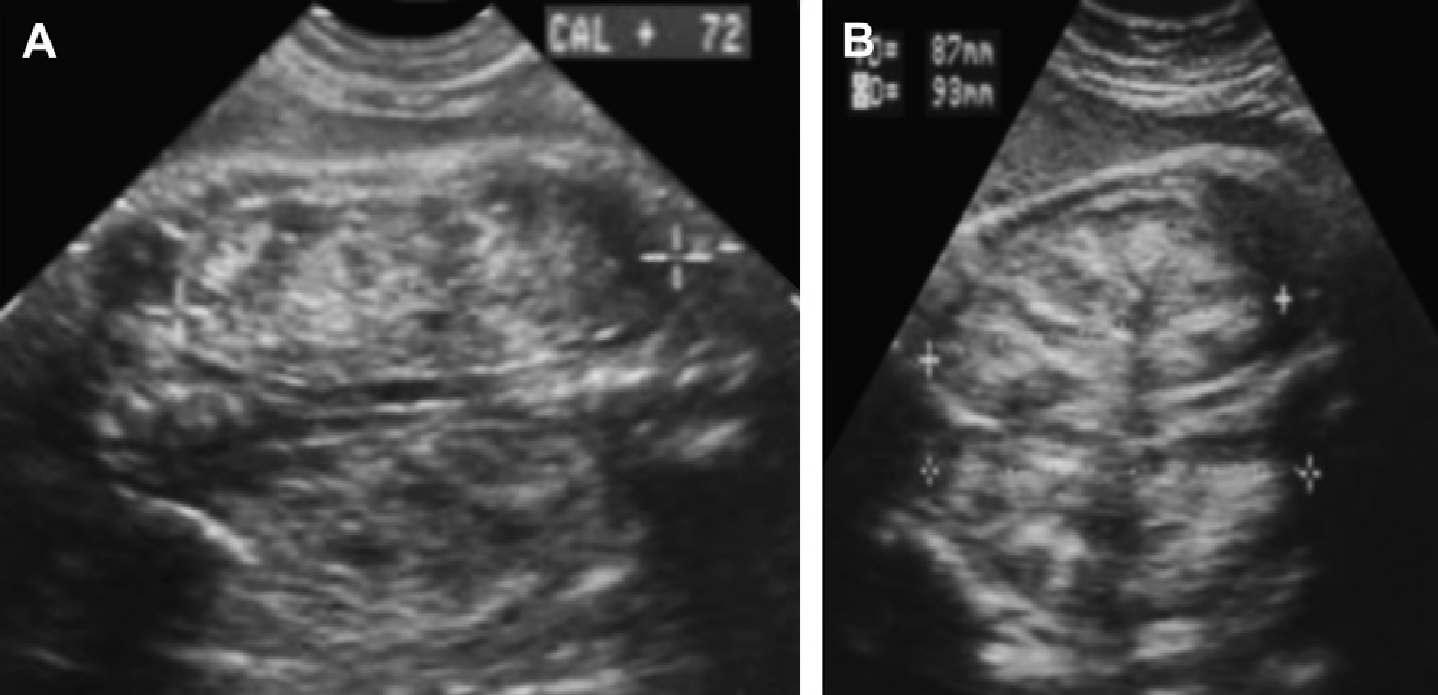
Рис. 21. АРПН, зміна структури під час вагітності. (А) Акушерська ультрасонограма на 30 тижні гестації показує, що обидві нирки значно розширені і гіперехогенні. (В) Акушерська ультрасонограма на 36 тижні гестації у того ж пацієнта. В даний час визначаються докази периферичного гіпоехогенного краю з виразною медулярною гіперехогенністю.
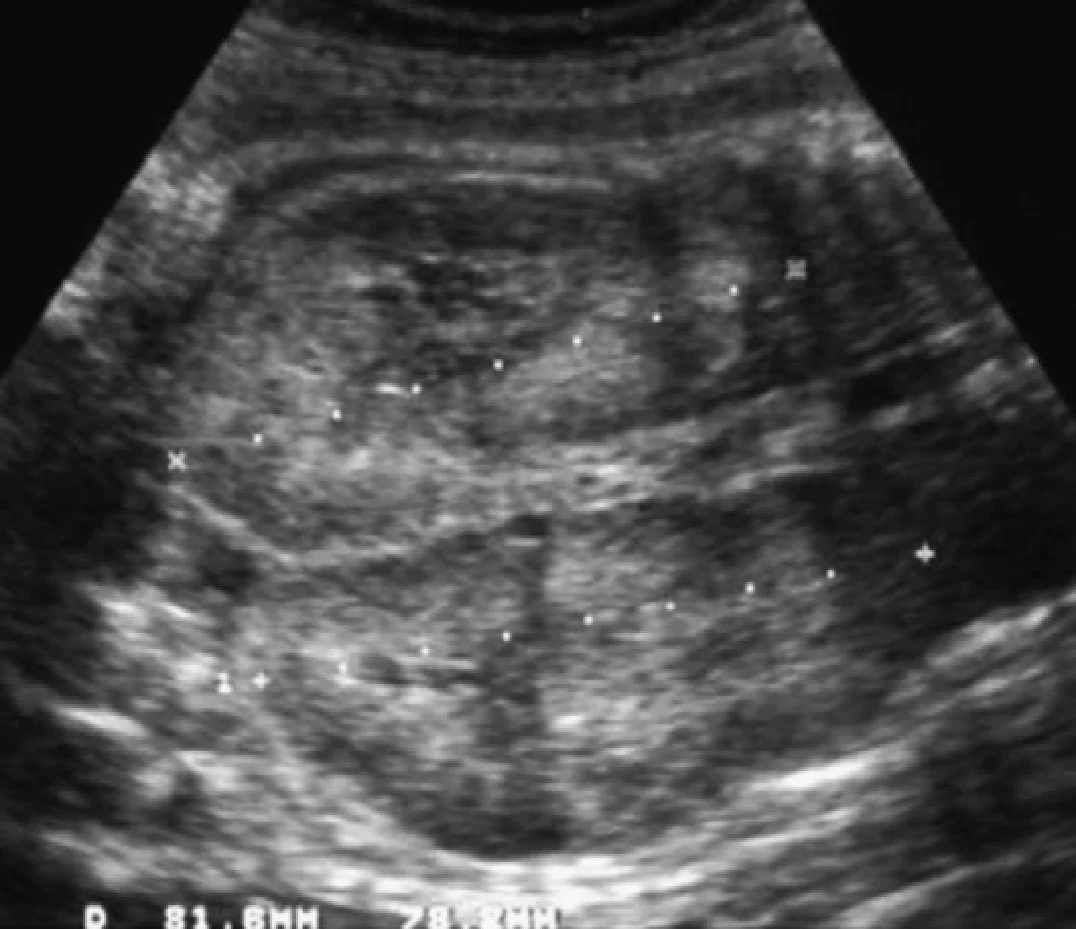
Рис. 22. АРПН, ультрасонограма в третьому триместрі. Обидві нирки мають розмір 8 см і периферичний гіпоехогенний ободок, який оточує гіперехогенну паренхіму.
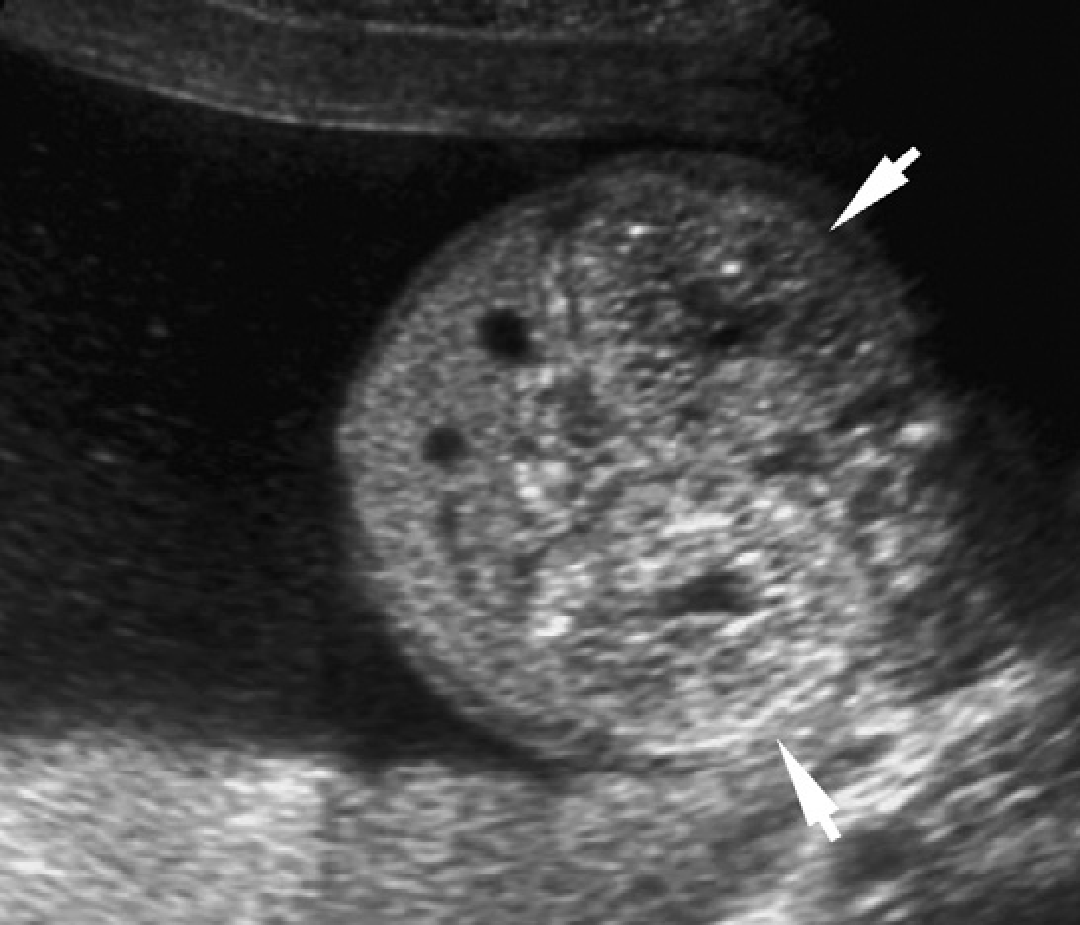
Рис. 23. Ранній важкий фетальний АРПН. Акушерська ультрасонограма на 19 тижні гестації показує великі недиференційовані нирки, які заповнюють більш ніж на 50% живота плода. Батьки обрали переривання вагітності. Патологія узгоджується з АРПН.
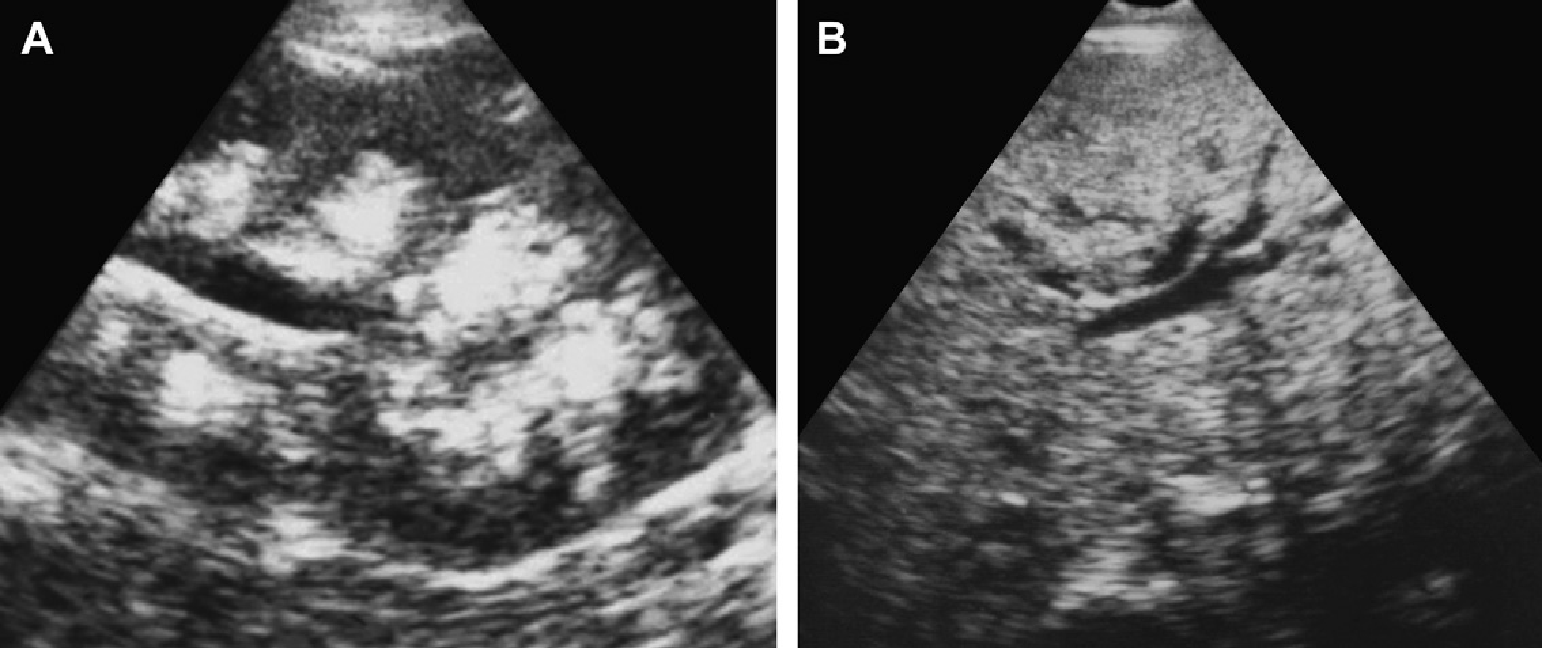
Рис. 24. АРПН, мозковий зразок у 2-річної дівчинки. (А) Ультрасонограма нирок показує виразно гіперехогенну мозкову речовину (псевдонефрокальциноз) в помірно збільшеній нирці. (В) гіперехогенну печінку з біліарною ектазією.
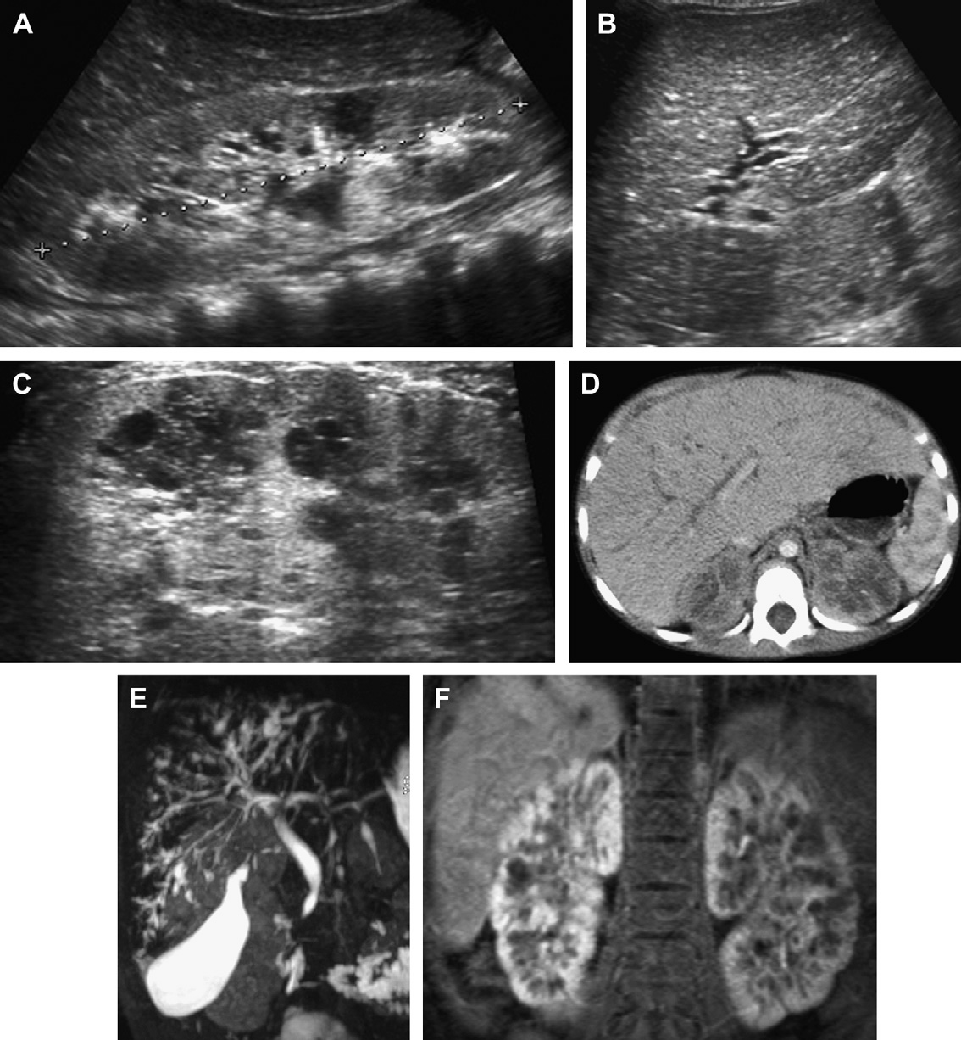
Рис. 25. АРПН, 2-дослідження річного пацієнта з приводу лихоманки невідомого походження. Різні методи візуалізації (ультрасонографія, КТ, МРТ) показують поєднане ниркове та печінкове ураження АРПН. При біопсії печінки виявлено кишкову паличку. (A) Поздовжня ультрасонограма правої нирки показує подовжену ехогенну нирку. (В) Ультрасонограма печінки показує дифузно ехогенну і грубозернисту паренхіму з біліарною ектазією. (С) Ультрасонограма нирки з високою роздільною здатністю показує численні дрібні мозкові кісти. (D) Контрастне КТ показує печінкову біліарну ектазію і слабке посилення ниркової кори. (Е) Коронарна Т2-зважена МРТ показує виразну ектазію біліарної системи. (F) Коронарна T2-зважена МРТ з контрастним посиленням показує множинні малі мозкові ниркові кісти і порушення архітектоніки.
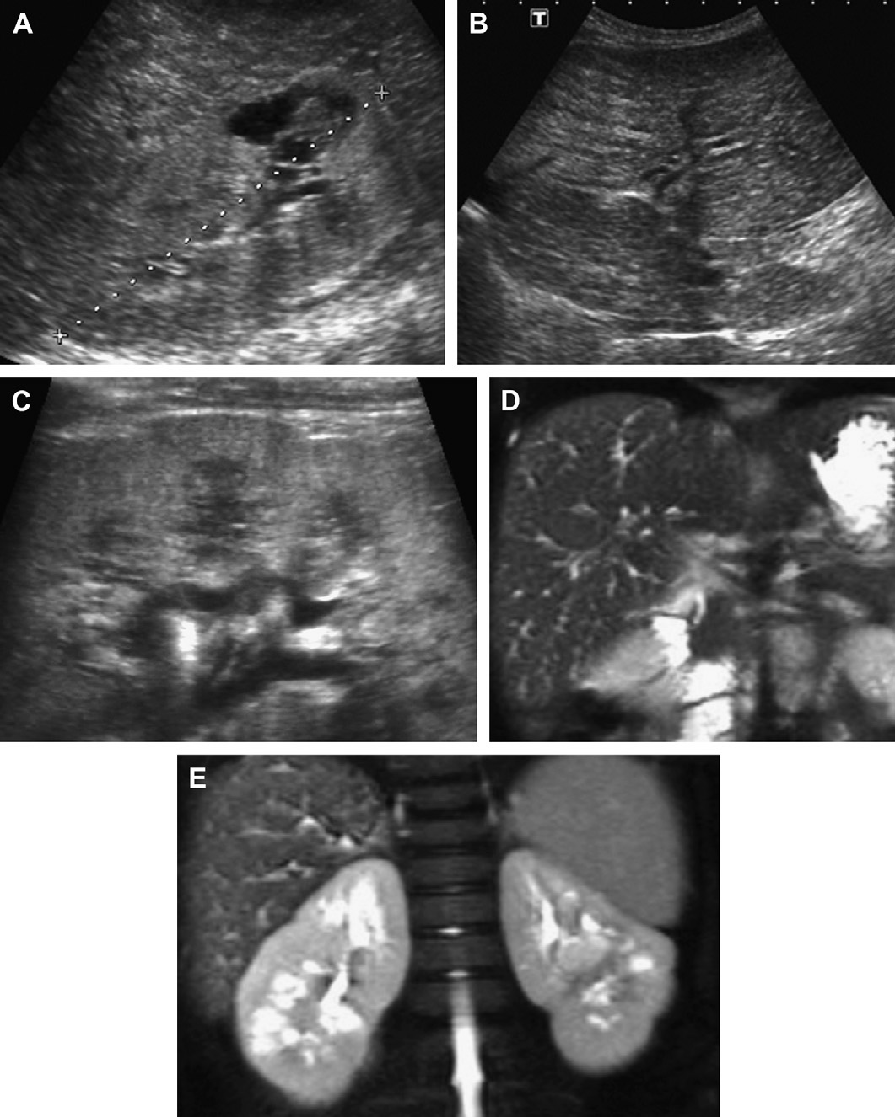
Рис. 26. АРПН, пацієнт у віці 18 місяців з лихоманкою неясного генезу тривалістю кілька тижнів. При біопсії печінки виявлено кишкову паличку. (A) Поздовжня ультрасонограма правої нирки показує ехогенну нирку з поганою кортико-медулярною диференціацією і кісти кіркової і мозкової речовини нижнього полюса. (В) Ультрасонограма печінки показує дифузну ехогенну і грубозернисту паренхіму з біліарною ектазією. (С) Ультрасонограма нирки з високою роздільною здатністю показує тонкі мозкові кістозні зміни. (D) Коронарна T2-зважена МРТ показує незначну ектазію біліарної системи. (Е) Коронарна Т2-зважена МРТ показує мозкові ниркові кісти.
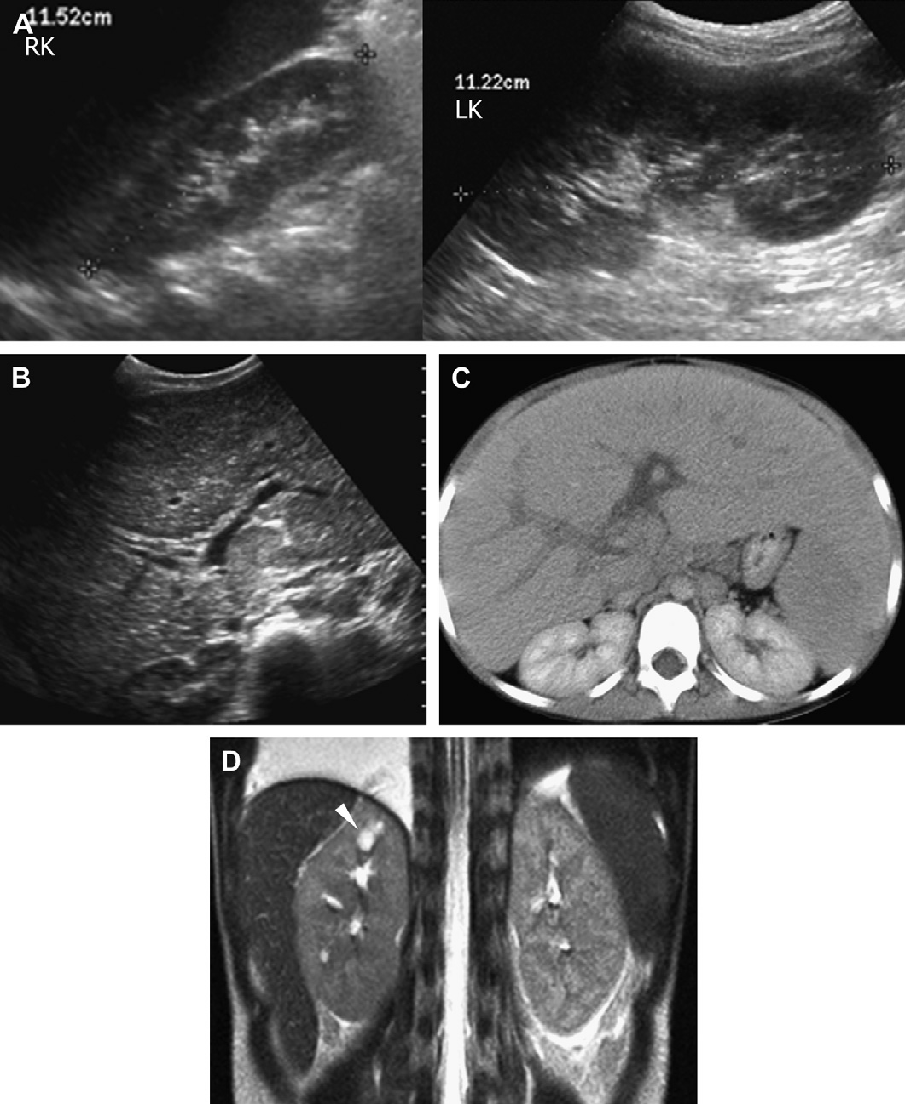
Рис. 27. АРПН, дослідження 10-річної дитини у зв’язку із загальним нездужанням, лихоманкою і діареєю після 1-тижневої відпустки в Домініканській Республіці. При біопсії печінки виявлено сальмонели. (А) Ультрасонограма нирок показує злегка збільшені нирки. Не виявлено ні кіст, ні порушень ехоструктури паренхіми. (B) Печінка збільшена, гіперехогенна з мінімальною біліарною ектазією. (С) КТ з контрастним посиленням показує збільшення печінки та біліарної ектазії; нирки без патології. (D) коронарне T2-зважене МРТ показує мозкову кісту (наконечник стрілки) в верхньому полюсі правої нирки.
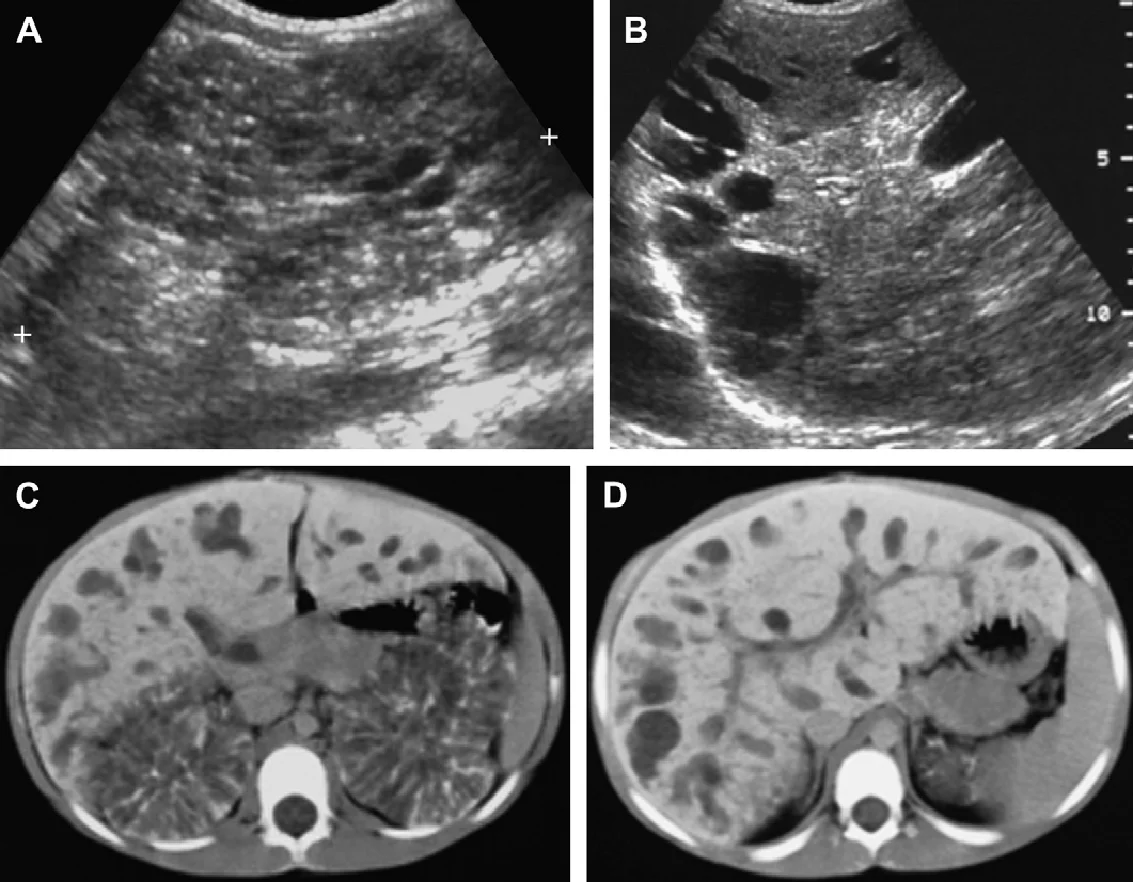
Рис. 28. АРПН; 9-річний пацієнт з тяжким ураженням обох нирок і печінки. (A) Поздовжня ультрасонограма збільшеної лівої нирки (курсори) показує кістозні і гіперехогенні зміни у всій паренхімі. (В) Ультрасонограма печінки показує біліарну ектазію і периферичні біліарні кісти. (С) КТ з контрастним посиленням показує збільшені нирки з покресленою трубчастою нефрограмою. (D) КТ печінки з контрастним посиленням показує біліарні кісти, які корелювали з даними ультрасонограми. Ознак портальної гіпертензії не виявлено (звичайний розмір селезінки).
Клінічні особливості та патогенез захворювання
Початок захворювання може не мати клінічних проявів. Погіршення тубулярної концентраційної функції викликає поліурію і полідипсію – звичайні симптоми нефронофтізіса. Прогресування до термінальної ниркової недостатності відбувається в різному віці, в залежності від форми нефронофтізіса. При ювенільній формі нефронофтізіса, найбільш часто зустрічається і обумовлена мутаціями в NPHP1, поліурія і полідипсія в 4-6 річному віці, а термінальна стадія ниркової недостатності розвивається до 13 років (середній вік). При дитячій та підлітковій формах нефронофтізіса, ниркова недостатність розвивається від 1 року до 3 років, і 19 років, відповідно.
Екстраренальні ураження (офтальмологічні, неврологічні, скелетні і печінкові) спостерігається в 20% випадків нефронофтізіса, деякі синдроми мають ознаки нефронофтізіса або пов’язані з NPHP генною мутацією (вставка 2).
2 Вставка
Синдроми, пов’язані з мутаціями NPHP генів
– Senior-Loken – пігментна ретинопатія
– Cogan- окуломоторна апраксія
– Joubert Тип B – недорозвинення хробака мозочка
– Saldino-Mainzer – конусоподібний епіфіз
– Sensenbrenner – краніоектодермальная дисплазія
– Jeune – короткі ребра
– Rhyns – гіпопітуїтаризм, пігментна ретинопатія, скелетна дисплазія
– Boichis проліферація біліарних протоків і фіброз печінки
– Alstrom – дистрофія сітківки, втрата слуху, ожиріння, цукровий діабет 2 типу- Meckel-Gruber – енцефалоцеле, полідактилія, кістозні нирки
Медулярний полікістоз, аутосомно-домінантний стан, відрізняється від нефронофтізіса пізнішим віком виникнення уремії (в зрілому віці) і відсутністю екстраренальних проявів (крім подагри).
Сонографічні особливості
Ультрасонограма може бути нормальною на ранніх стадіях захворювання (рис. 29-31). Втрата кірково-медулярної диференціації і гіперехогенність паренхіми часто спостерігається на тлі нирок нормальних розмірів. Кісти нирок (медулярної або кірково-медулярної локалізації) стають видимими, коли у пацієнтів розвивається термінальна стадія ниркової недостатності. Приблизно у 25% пацієнтів не виявляються кісти під час гістологічного дослідження або на ультрасонограмах. Найбільш характерна картина на ультрасонограмах при нефронофтізісі до початку діалізу – декілька мозкових кіст в нирці, практично нормального розміру, з відсутністю диференціації (що не характерно в термінальній стадії ниркової патології).
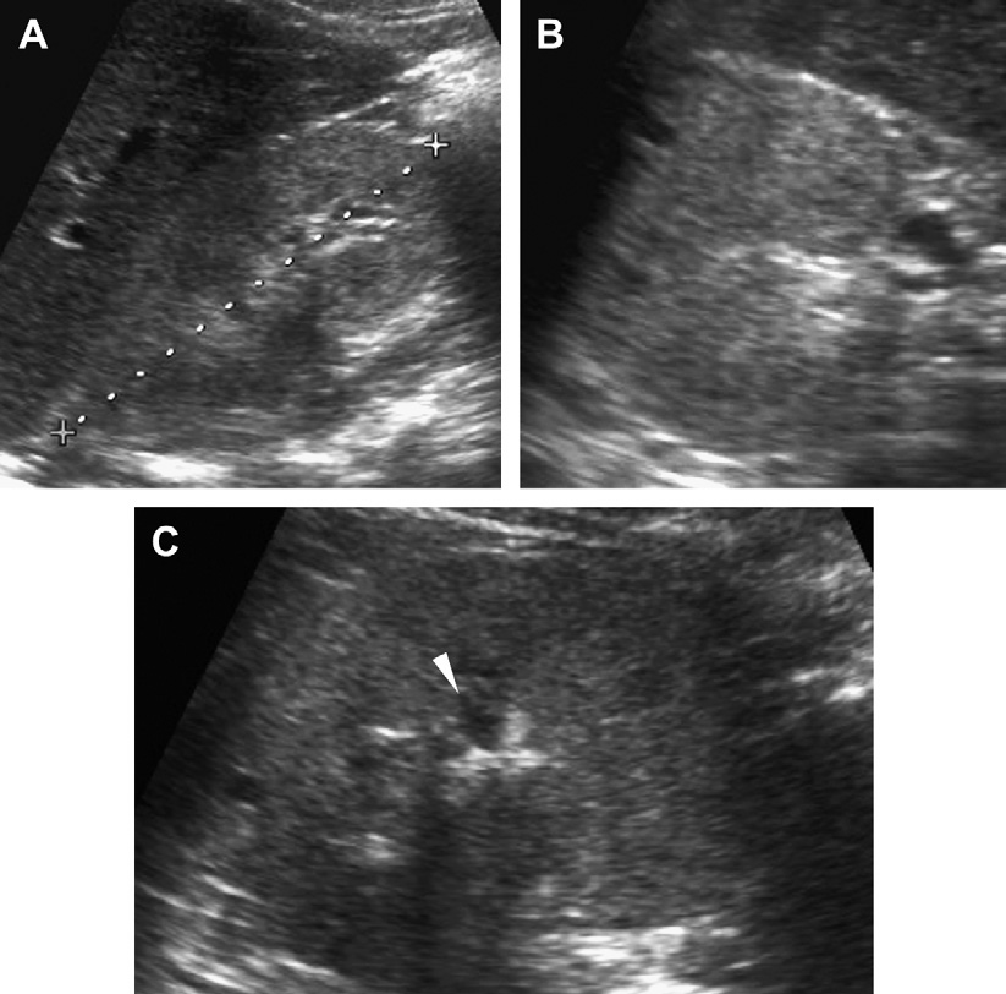
Рис. 29. Нефронофтізіс, синдром Cogan у 12-річного пацієнта з хронічною нирковою недостатністю. (A) Поздовжня ультрасонограма показує праву нирку (курсори) розміром 9,4 см, з повною відсутністю кірково-медулярної диференціації і гіперехогенною корою. (В) Поперечне зображення правої нирки показує невелику кісту в зоні кірково-медулярного переходу. (C) Поздовжня ультрасонограма лівої нирки також показує мозкову кісту (наконечники стрілки).
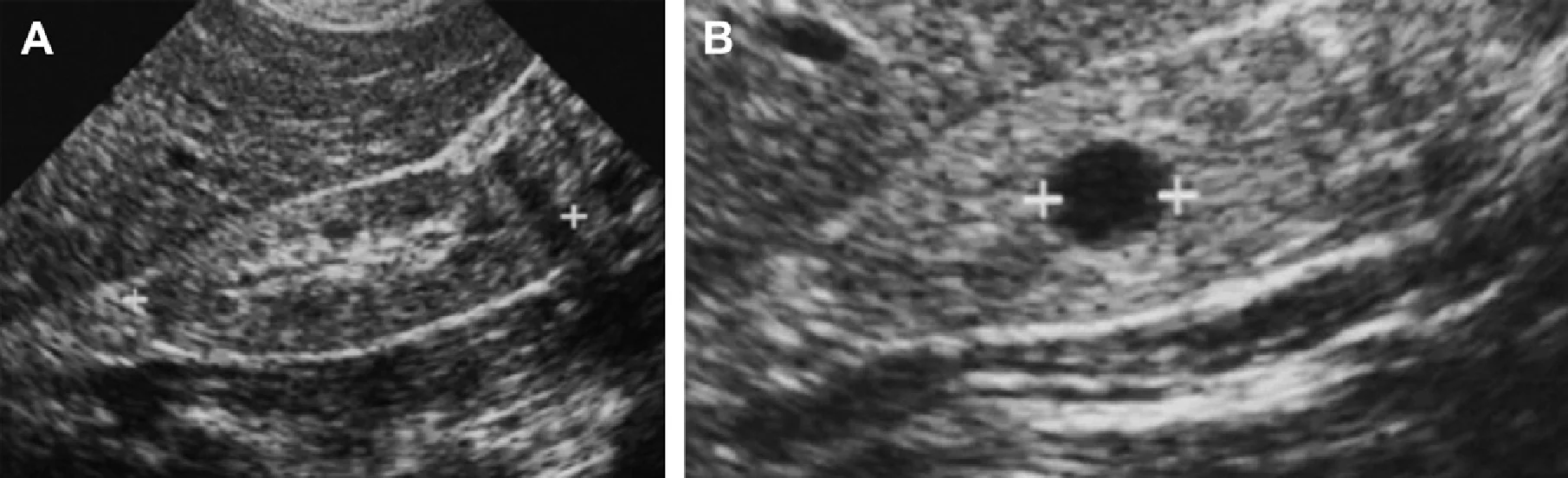
Рис. 30. Нефронофтізіс; Senior-Loken у 12-річного пацієнта в термінальній стадії ниркової недостатності. (A) Поздовжня ультрасонограма показує праву нирку (курсори) розміром 8 см, з втратою кірково-медулярної диференціації. (B) Одинична відособлена медулярна кіста (курсори) в правій нирці.
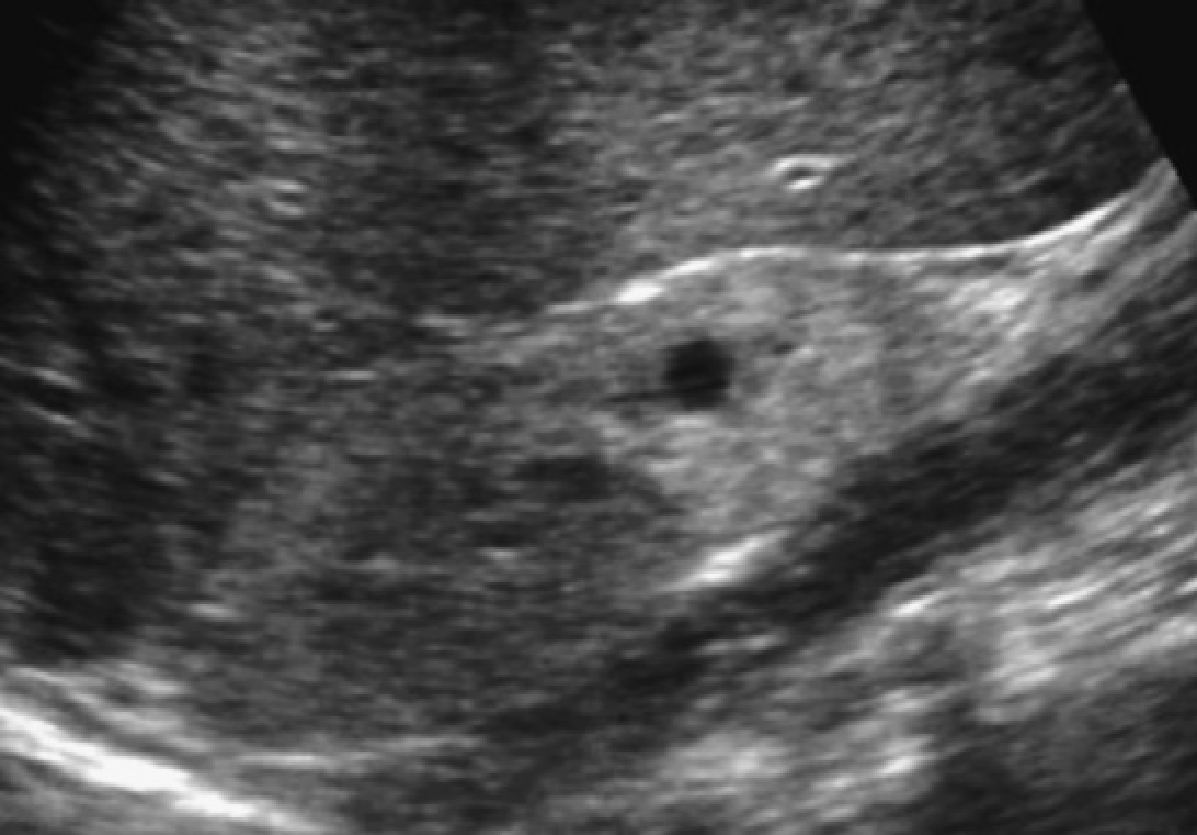
Рис. 31. Нефронофтізіс; 10-річний пацієнт перед трансплантацією нирки. Поздовжня ультрасонограма правої нирки показує одиничну мозкову кісту в гіперехогенній нирці.
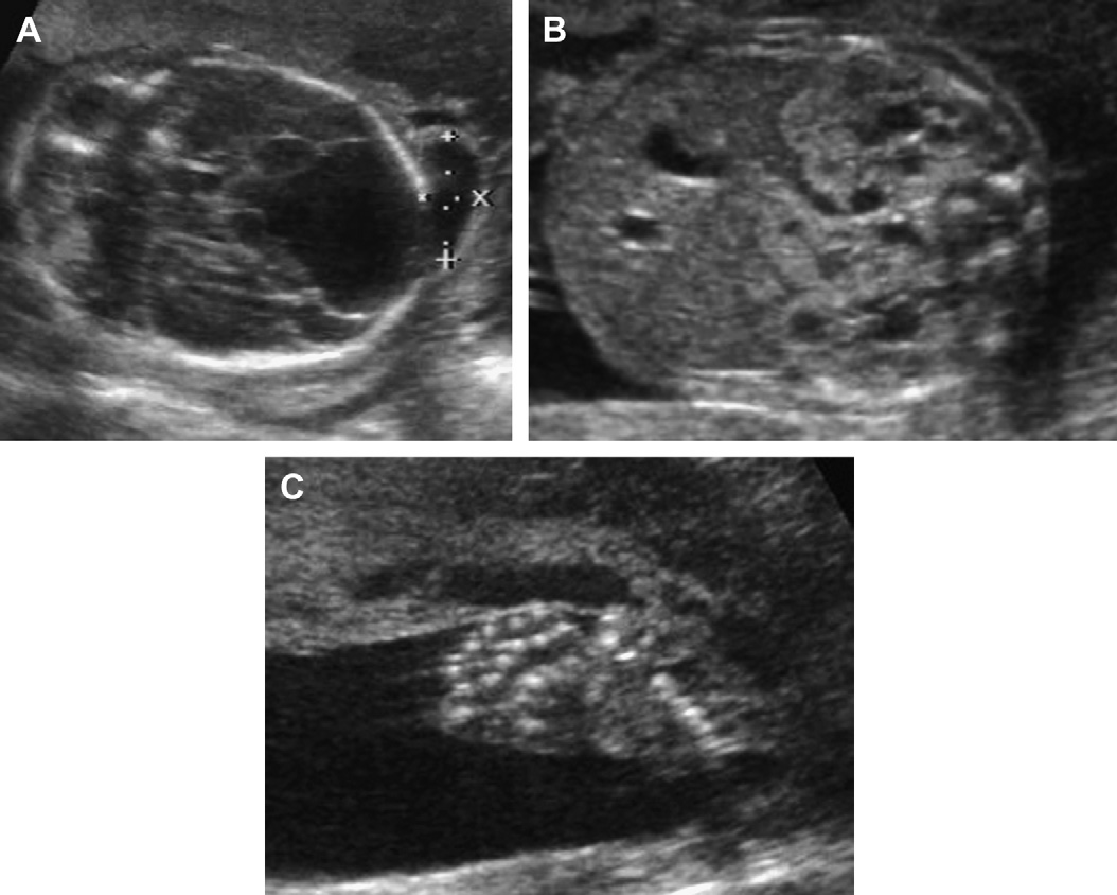
Рис. 32. Синдром Meckel-Gruber (летальний випадок). Акушерська ультрасонограма на 19 тижні гестації. (А) Аномальна задня черепна ямка з цефалоцелє (курсори). (B) Двосторонні ниркові кістозні мозкові аномалії. (C) Полідактилія.
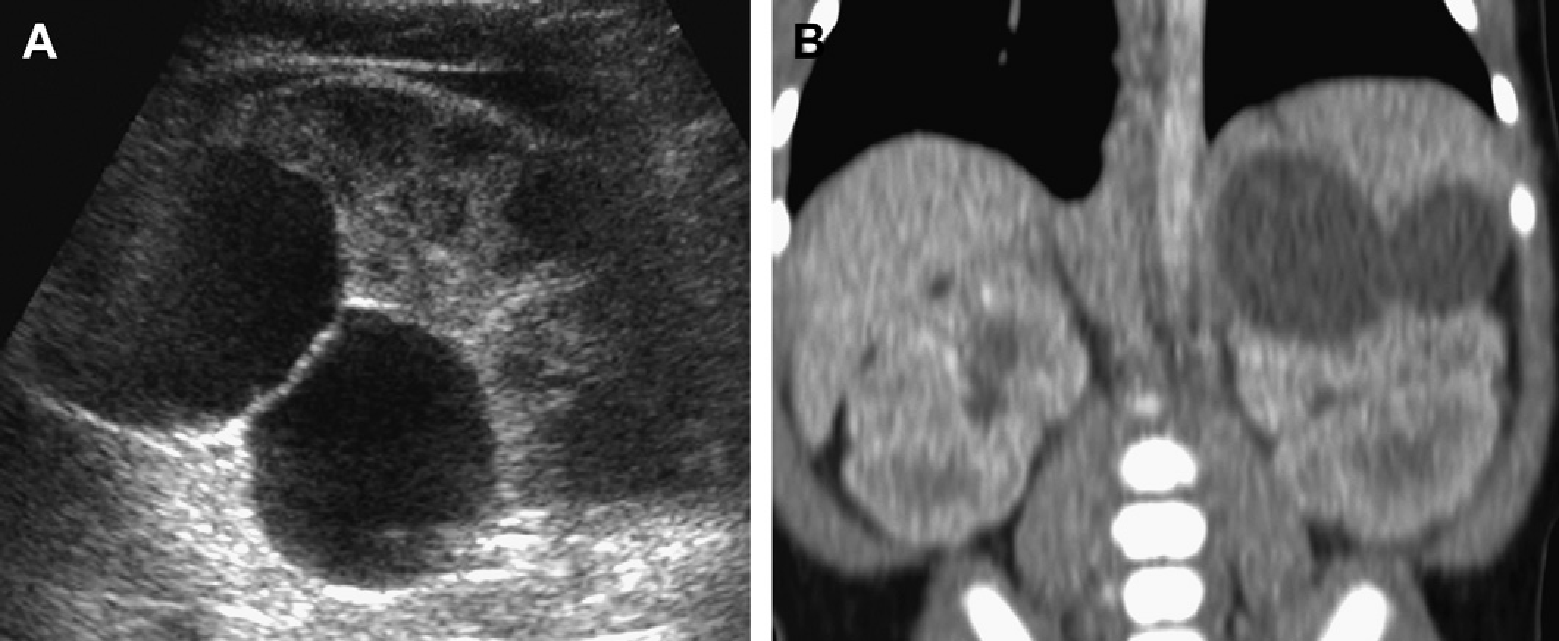
Рис. 33. Синдром Беквіт-Відемана у 14-денного немовляти. (А) Поздовжня ниркова ультрасонограма показує макрокісти в лівій нирці. (В) Реформатоване коронарне КТ показує великі кісти в верхньому полюсі лівої нирки.
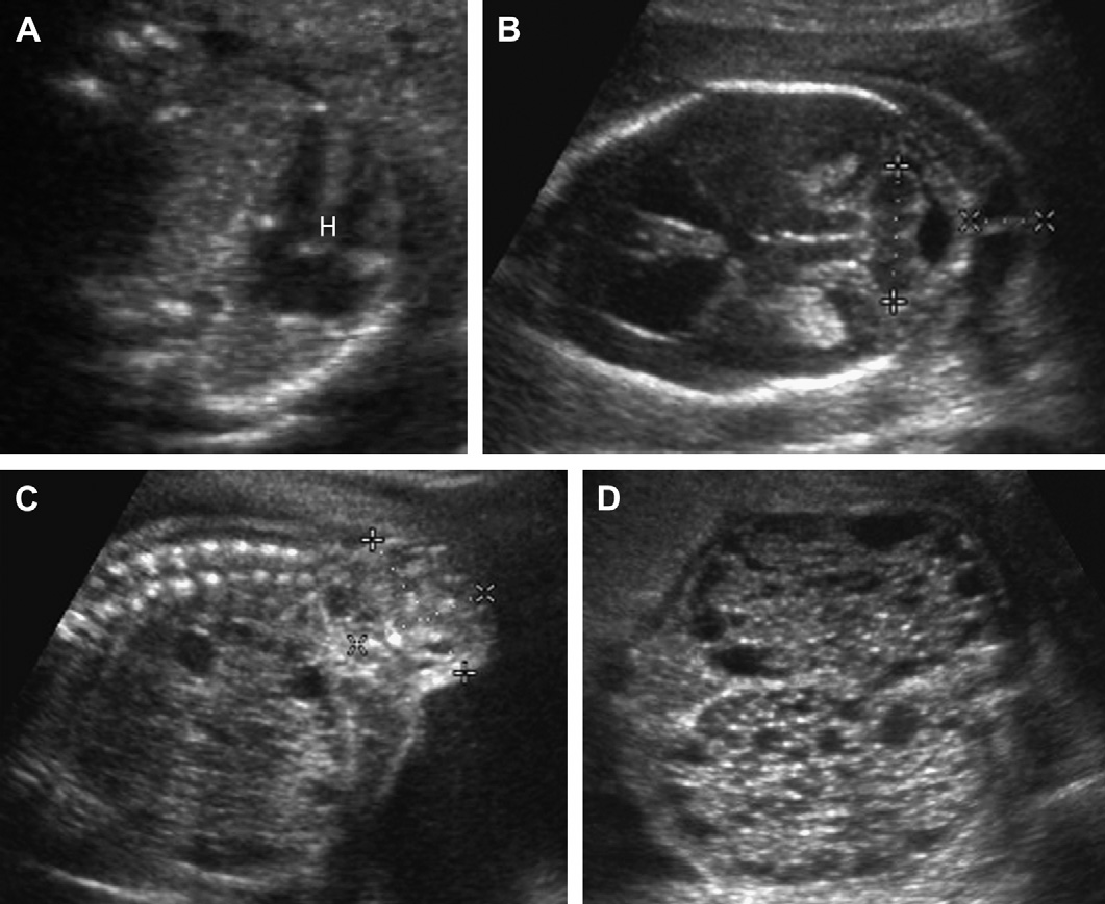
Рис. 34. Синдром Фрінса (летальна форма). Акушерська ультрасонограма на 19 тижні гестації. (А) Поперечна чотирьохкамерна проекція через грудну клітку показує серце (H), яке зміщене вправо діафрагмальною грижею. (B) Осьове зображення голови показує гідроцефалію і потиличну кістозну гігрому. (С) Поздовжнє зображення дистального відділу хребта показує куприкову тератому (курсори). (D) Обидві нирки збільшені з дифузними важкими кістозними змінами.
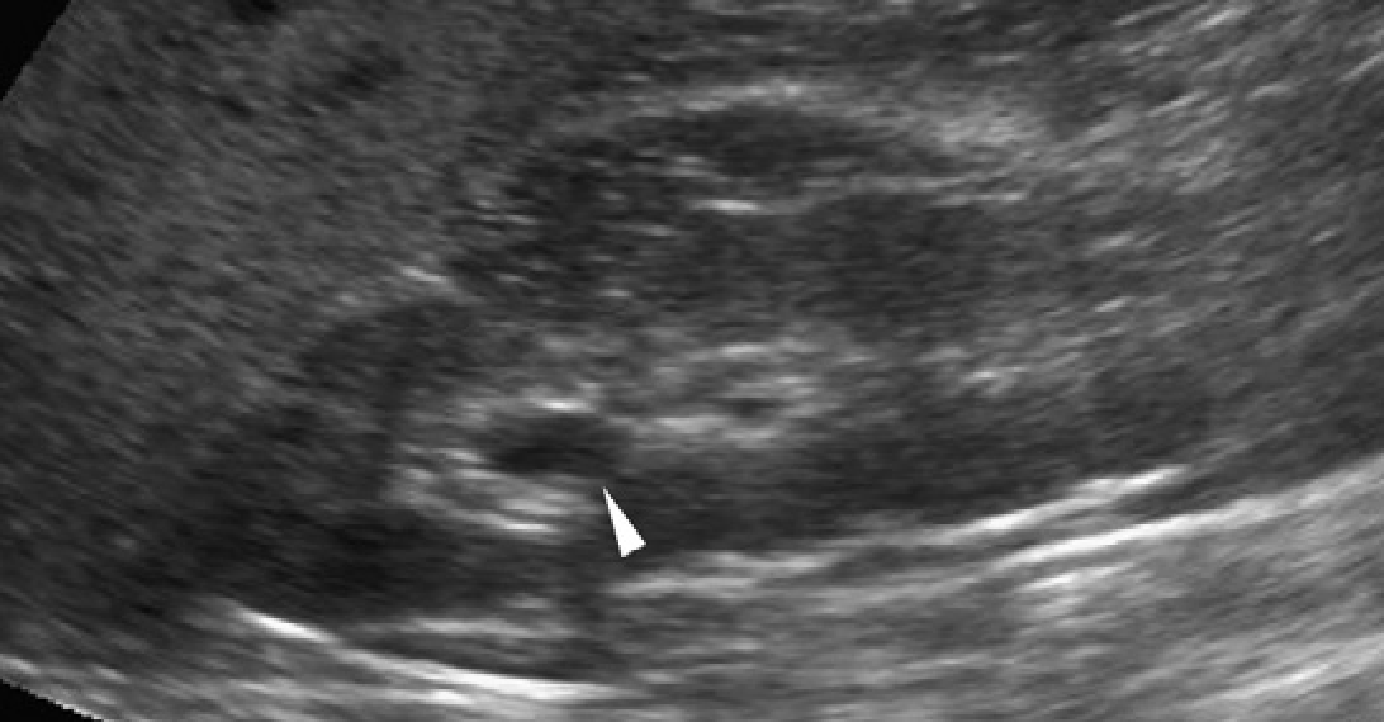
Рис. 35. Синдром Барді-Бідля. Поздовжня ультрасонограма нирок у 10-річного пацієнта показує повну втрату кірково-медулярної диференціації. Візуалізується поодинока медулярная кіста (наконечник стріли).
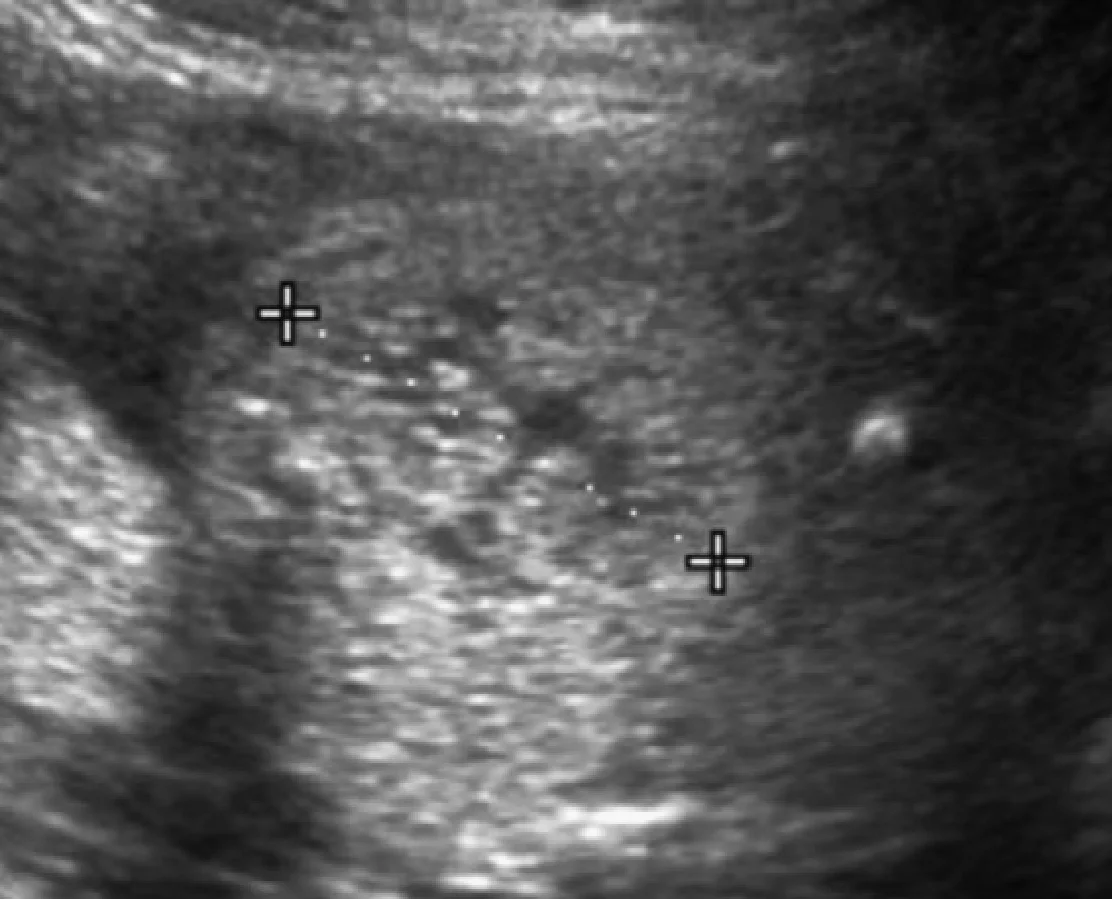
Рис. 36. Синдром Барді-Бідля; пренатальна діагностика. Акушерська ультрасонограма на 21 тижні гестації показує збільшену до 4 см з аномальною ехогенністю нирку. Також визначається полідактилія (не показано). Батьки вибрали переривання вагітності.
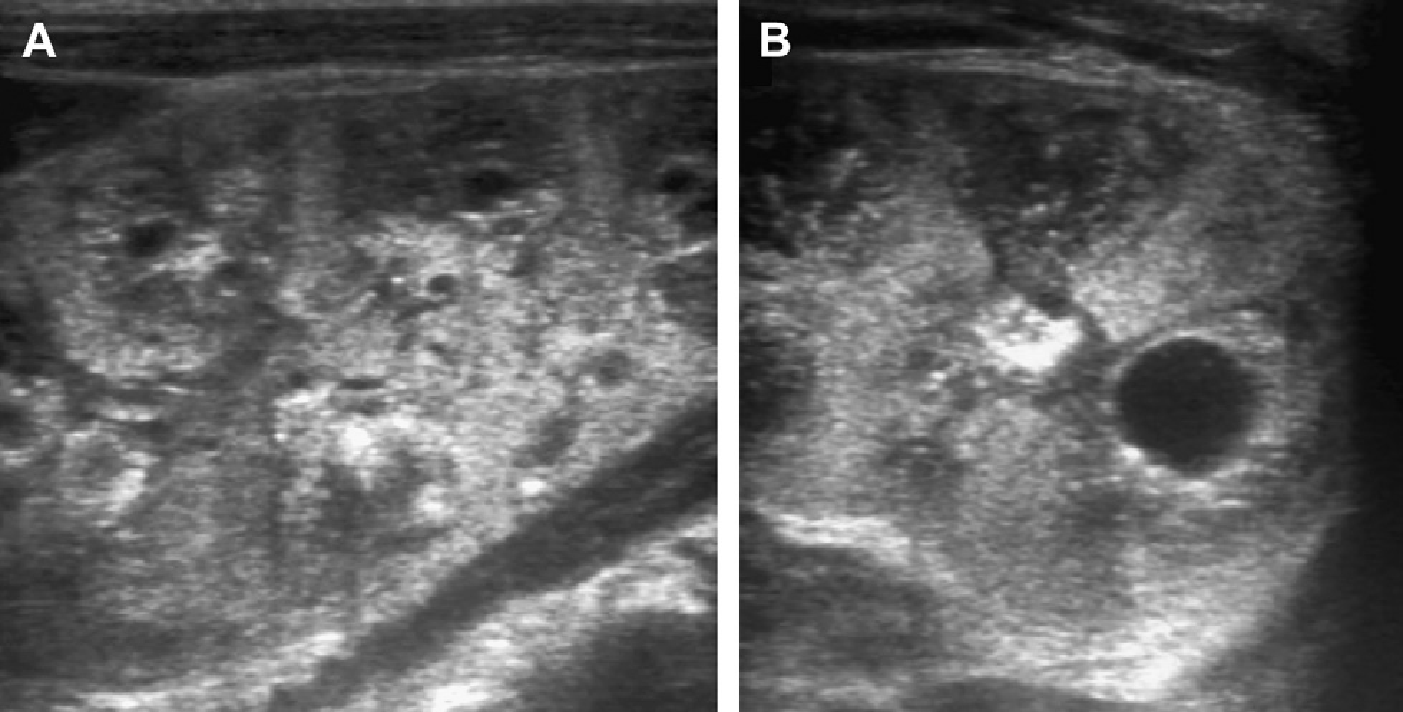
Рис. 37. Триденний немовля з аномалією 10 хромосоми. Ультрасонограма правої нирки з високою роздільною здатністю (А) і лівої нирки (В) показує великі мозкові кістозні зміни.
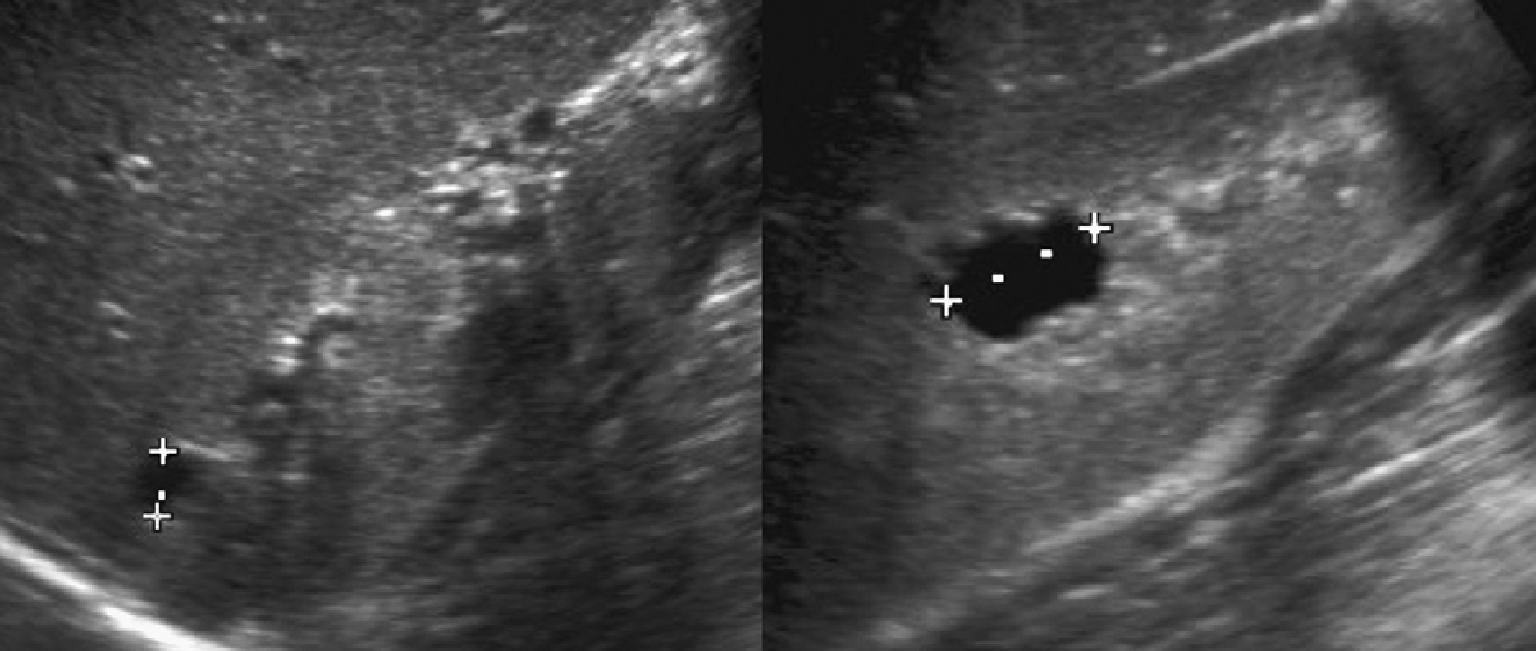
Рис. 38. Синдром Вільямса, 14-річний пацієнт з однією правою ниркою. Кісти визначаються в кірковій і мозковій речовині (курсори).
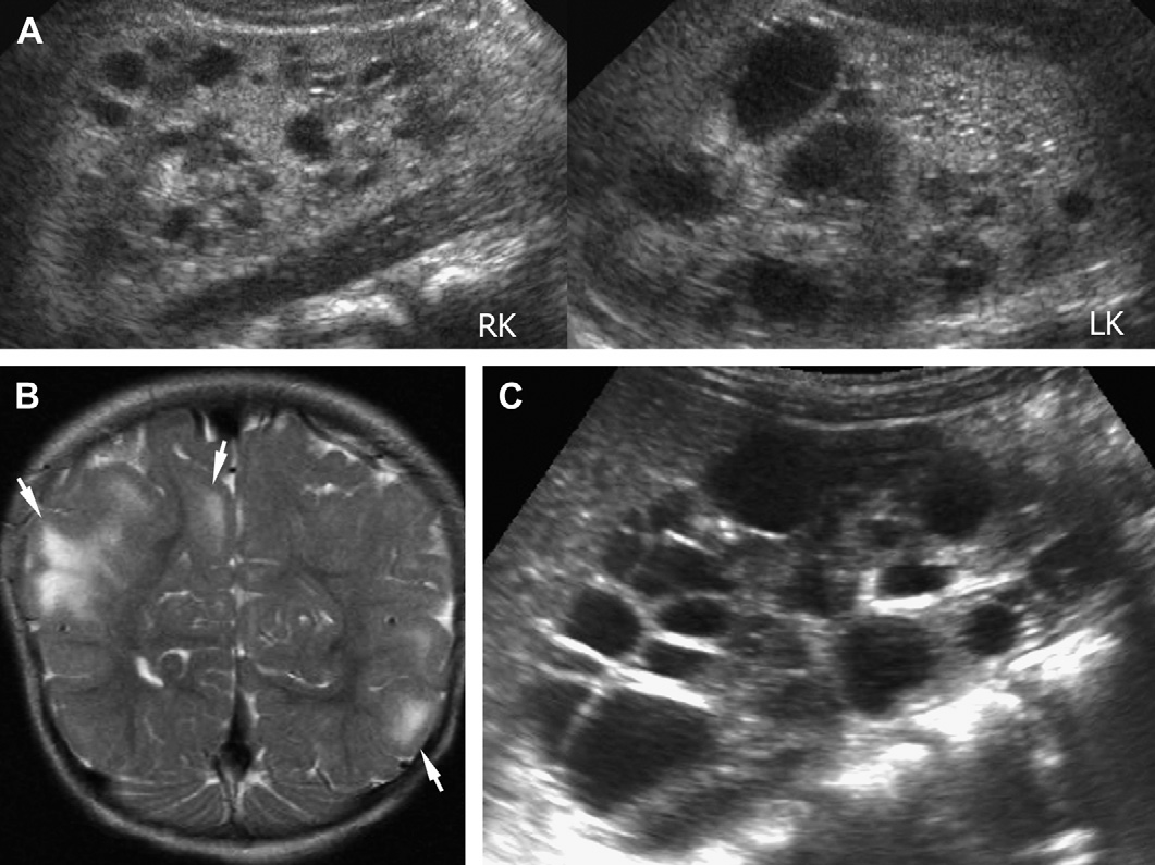
Рис. 39. Туберозний склероз. Післяпологове дослідження двосторонніх кіст нирок, які визначалися в період внутрішньоутробного розвитку. (А) Ультрасонограма нирок в 1 місяць життя показує наявність двосторонніх кіст, як в кірковій, так і мозковій речовині. (В) МРТ головного мозку в 10-місячному віці. Коронарне T2-зважене зображення показує двосторонні субкортикальні гамартоми (стрілки), що характерно для туберозного склерозу. (C) Ультрасонограма нирок у віці 2 років показує прогресивний розвиток кіст.
ГЛОМЕРУЛОКІСТОЗНІ ЗАХВОРЮВАННЯ НИРОК
Нозологія
Термін «гломерулокістозний» відноситься до клубочкових кіст (розширення простору Боумана). Гломерулярні кісти не специфічні для однієї патології. Гломерулокістозні захворювання являють собою первинні захворювання; гломерулокістозні нирки – це нирки з клубочковими кістами в якості домінуючого прояву, але різноманітними по етіології (наприклад, синдроми вад розвитку, такі як: склероз туберози, рото-пальце-лицьовий дизостоз тип I).
Генетика
Більшість гломерулокістозних захворювань нирок (ГКЗП) передаються відповідно за аутосомно-домінантним типом успадкування і виявляються у немовлят або в контексті сімейної історії АДПН (ГКЗП новонароджених з АДПН фенотипу), або без сімейного анамнезу (спорадичні ГКЗП новонароджених). Клінічні прояви ГКЗП можуть розвиватися у дітей старшого віку і дорослих, з наявністю сімейного анамнезу (домінуючі), так і спорадичні випадки, що відображають появу нових мутацій. Був описаний зв’язок ГКЗП з мутаціями гена 1B ядерного фактора гепатоцитів, також в деяких сім’ях знайдений зв’язок з раннім терміном прояву діабету підлітків тип 5. Вади розвитку статевих шляхів також реєструються при HNF1B мутаціях. ГКЗП є або домінуючими, або спорадичними, зустрічаються як у немовлят, так і у більш дорослих пацієнтів.
Сонографічні особливості
При ГКЗП варіанти АДПН у немовлят, нирки збільшені, гіперехогенні, без кортико-медулярної диференціації. Субкапсулярні кіркові кісти досить типові для ГКЗП. Кісти можуть розвиватися внутрішньоутробно або тільки після народження. При іншій формі ГКЗП, нирки можуть бути гіпоплазовані, мати нормальний розмір, або бути збільшеними. Гломерулокістозні нирки часто поєднуються з синдромами вад розвитку.
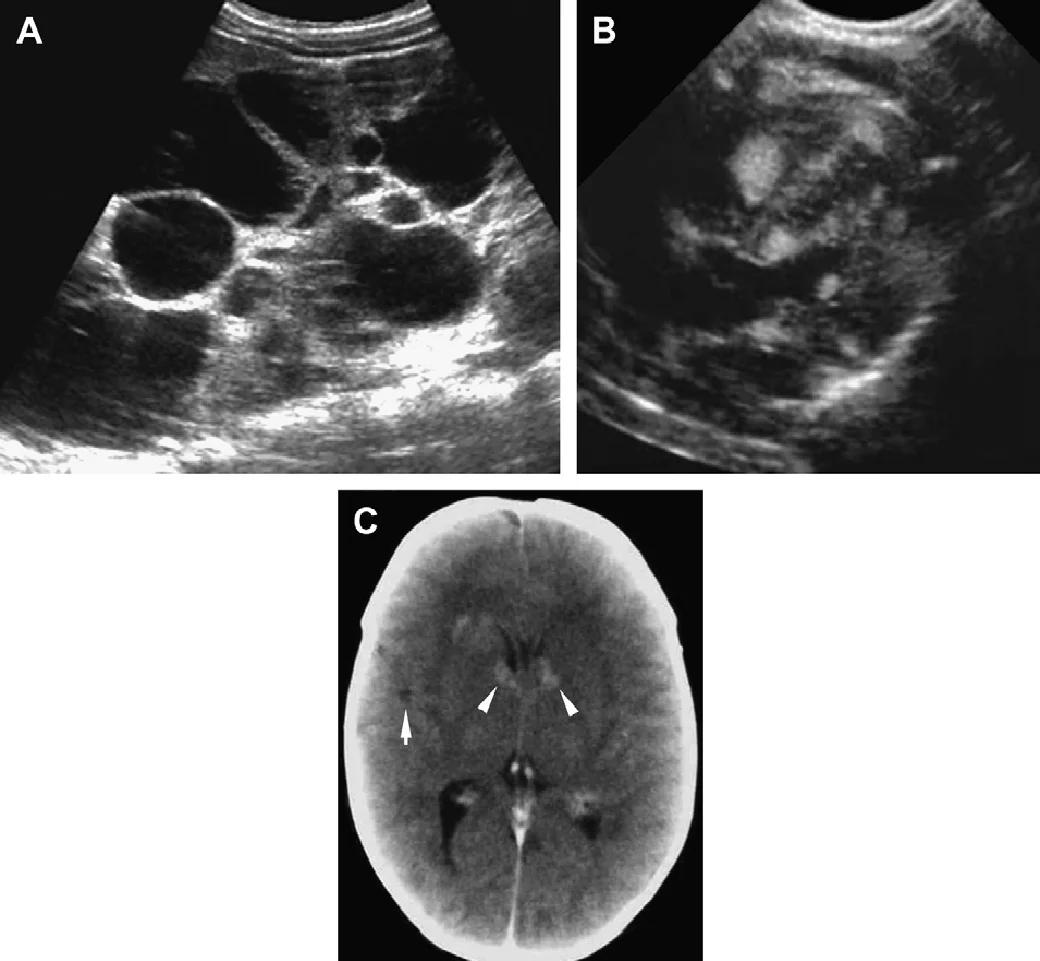
Рис. 40. Туберозний склероз. Дослідження новонародженого в зв’язку з даними пренатальної ультрасонограми, на якій виявлено кардіальна рабдоміома і ниркові кісти. (A) Поздовжня ультрасонограма правої нирки показує протяжні кістозні зміни. Ліва нирка має схожі ознаки. (В) Добре видно множинні осередки ехогенної кардіальної рабдоміоми. (С) КТ головного мозку з контрастним посиленням показує підкіркові гамартоми і ураження на рівні отвору Монро, що типово для туберозного склерозу.
СИНДРОМИ ВАД РОЗВИТКУ
Нозологія і генетика
Численні синдроми можуть мати клінічні прояви кіст нирок (3 Вставка).
3 Вставка
Вади розвитку на тлі полікістозу нирок
– Туберозний склероз
– Синдром Барді-Бідля
– Синдром Беквіт-Відемана
– Синдром Меккеля-Грубера
– Синдром Гіппеля-Ліндау
– Синдром Зельвегер
– Синдром короткі ребра, полідактилія
– Синдром Жуна
– Синдром Елліс-ван Кревелда
– Рото-пальцьо-лицевий дизостоз тип I
– Глутаровий ацидурия тип II
– Синдром Івмарка (нирково-печінкова-панкреатична дисплазія)
– Синдром Мардена-Уокера
– VACTERL
– Синдром Сміт-Лемлі-Опітц
– Синдром Аладжіля
– Синдром Етлера-Данлоса
– Синдром Тернера
– Трисомії (13-15, 18, 21, 10)
Їх можна поділити за їх типом успадкування (1) (аутосомно-домінантне [наприклад, склероз туберози, хвороба Гіппеля-Ліндау], аутосомно-рецесивне [наприклад, синдроми Меккеля-Грубер, Жуна, Барді-Бідля], Х-хромосома домінантне [рото-пальцьо-лицевий дизостоз тип I], хромосомні порушення [трисомії D, E, 21, синдром Тернера]); (2) по їх патологічним характеристикам: дифузна кістозна дисплазія, як при синдромі Меккеля-Грубера, Беквита-Видемана, глутарова ацидурія тип II, VACTERL, гломерулокістоз нирок, як при туберозному склерозі, рото-пальцьо-лицевий дизостоз тип I, синдром коротких ребер і полідактилії, синдром Жуна, Зеллвегер, трисомія 13; і (3) пов’язані з патологією ними клінічні прояви і перебіг. Деякі синдроми є летальними внутрішньоутробно (наприклад, синдром Меккеля-Грубера (Рис. 32), рото-пальцьо-лицевий дизостоз тип I у хлопчиків), деякі з них можуть бути пропущені в дитинстві і діагностуються лише в більш пізньому віці (наприклад, синдром Бардета-Бідля, який характеризується пігментного ретинопатію, аномаліями дистальних відділів кінцівок, аномаліями нирок, ожирінням, гіпогонадизмом у чоловіків, розумовою відсталістю), деякі виявляються клінічно тільки в зрілому віці, але можуть бути діагностовані за допомогою аналізу мутацій (пресимптоматичний тест) у членів сім’ї ( наприклад, хвороба фон Хиппеля-Ліндау).
Наявність супутніх вад розвитку (полідактилія, аномалії центральної нервової системи, надлишковий ріст, невеликі груди, вади серця і так далі) допомагає поставити попередній діагноз (наприклад, полідактилія в поєднанні з кістами нирок проявляється при синдромі Меккеля-Грубера, синдромі Барді-Бідля, синдромі коротких ребер і полідактилії, синдромі Сімпсона-Голабі-Бемеля). Серед різних синдромів вад розвитку (Рис. 33-38), полікістоз нирок при туберозному склерозі, є важливою ланкою для чіткого визначення патології (Рис. 39 і 40).
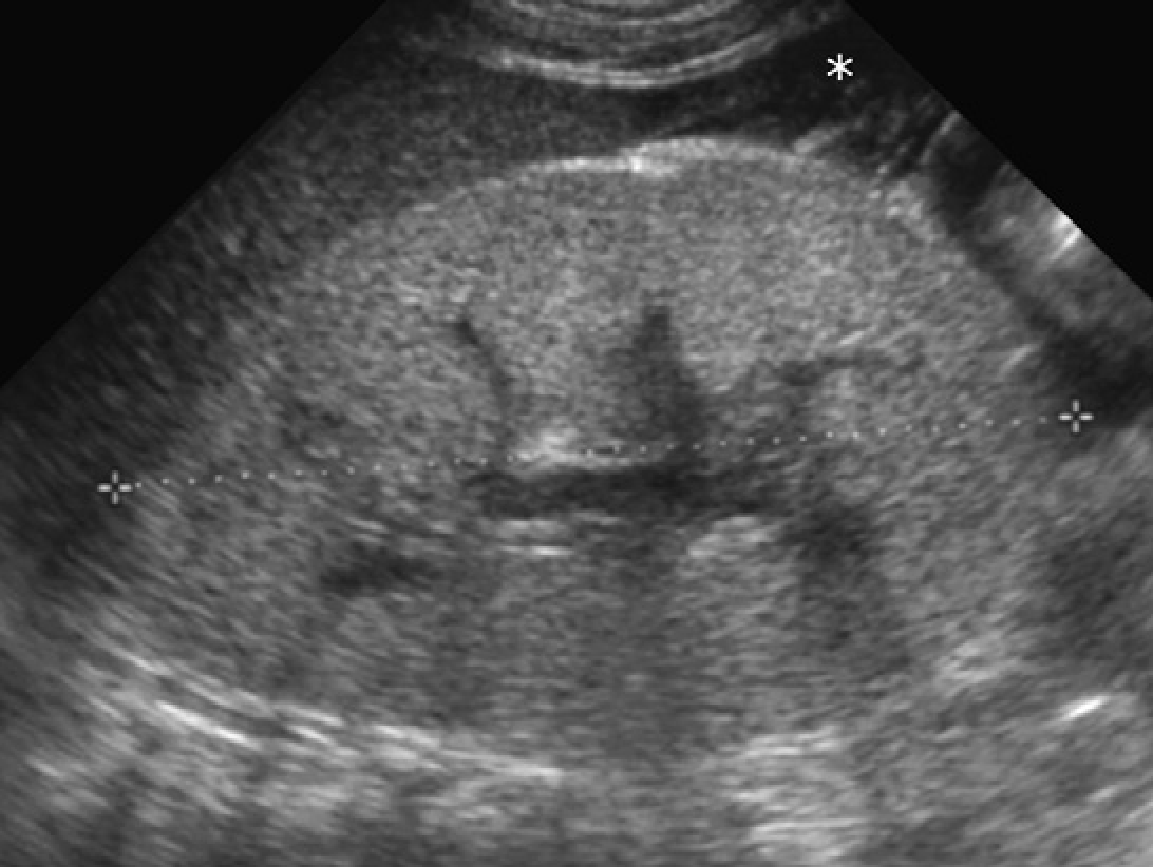
Рис. 41. Чотирьох-місячний пацієнт з швидко прогресуючим нефротичним синдромом. Ультрасонограма нирок показує велику (7,5 см) ехогенну нирку (курсори) з втраченою кірково-медулярною диференціацією і супутнім асцитом (зірочка). Перитонеальний діаліз було розпочато в 8 місячному віку, після чого проведена успішна трансплантація нирок.
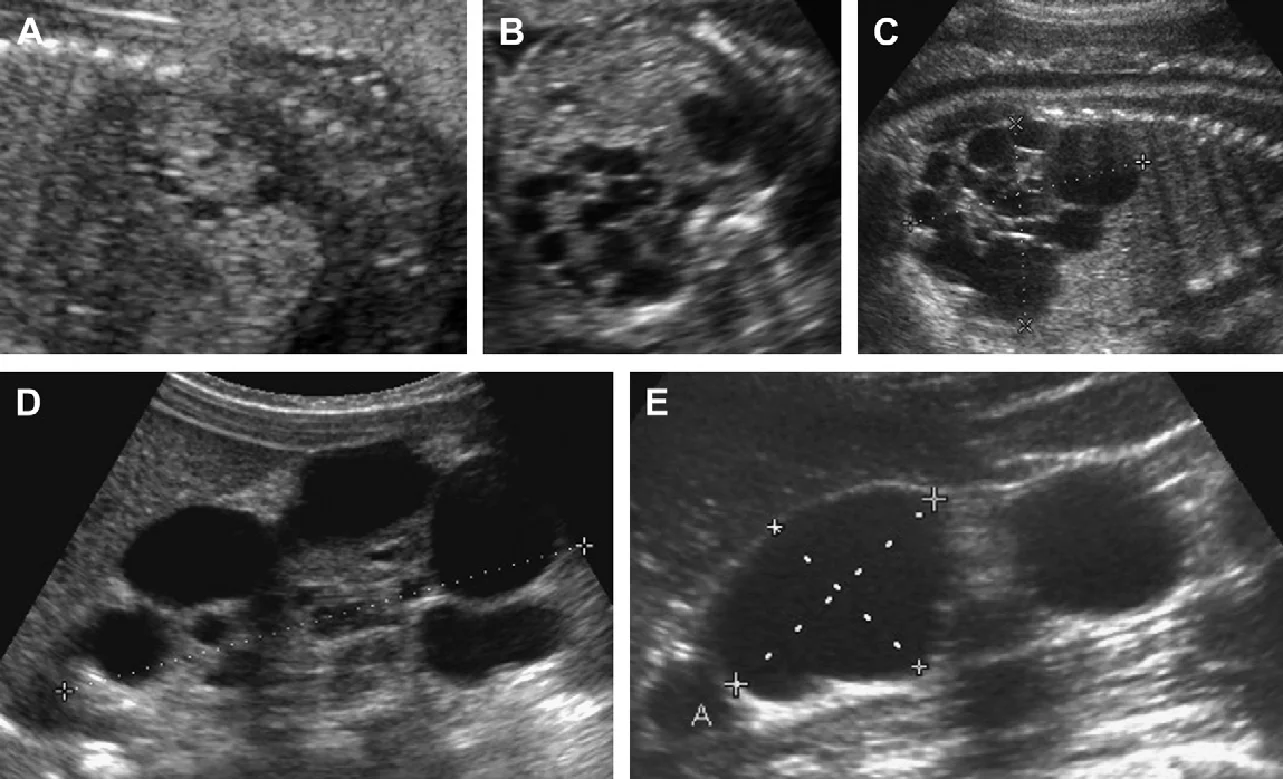
Рис. 42. Правосторонняя полікістозна дисплазія нирок. Нормальна ліва нирка (не відображено). Пренатальна і постнатальна картина для порівняння. Акушерська ультрасонограма на 16 тижні гестації (А), 20 тижні гестації (В), і 30 тижні гестації (С). Спочатку розташовані по периферії на 16 тижні гестації, розкиданими кісти стають випадковим чином на 30 тижні гестації. (D) Ультрасонограма правої нирки на 5-й день життя показує кісти різного розміру з тонкою проміжної перетинкою між ними і відсутністю нормальної паренхіми. (Е) Ультрасонограма нирок у віці 4-х років показує кісти в правій нирці.
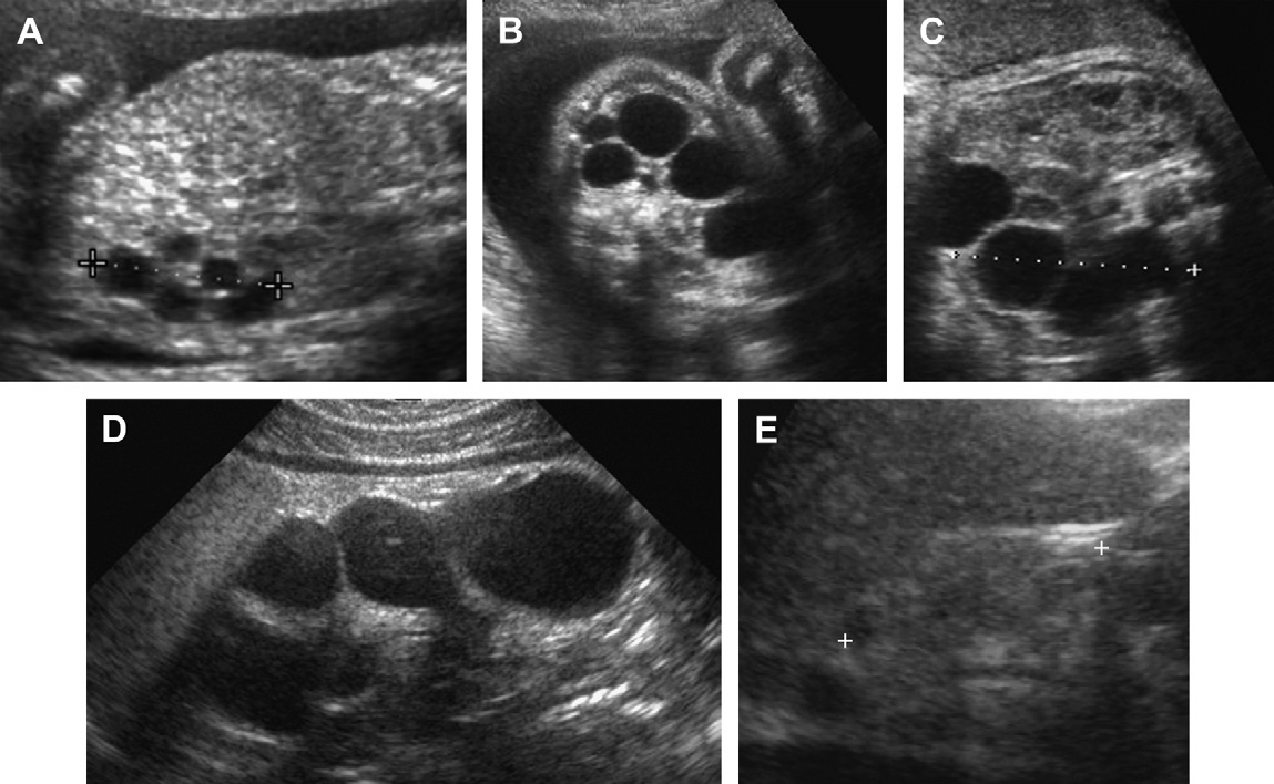
Рис. 43. Лівобічна полікістозна дисплазія нирок. Пренатальна і постнатальна картина. Акушерська ультрасонограма на 21 тижні гестації (А), 32-му тижні гестації (B), і 36 тижні гестації (С). Макрокісти нирки визначаються під час вагітності. (D) Ультрасонограма нирок в місячному віці показує ліву нирку довжиною 7 см і множинні кісти в ній, без ознак дилатації миски. Права нирка нормальна (не відображено). (Е) Ультрасонограма лівої нирки в 2 роки демонструє атрофічну нирку (4,5 см) (курсори) з майже повною елімінацією кіст.
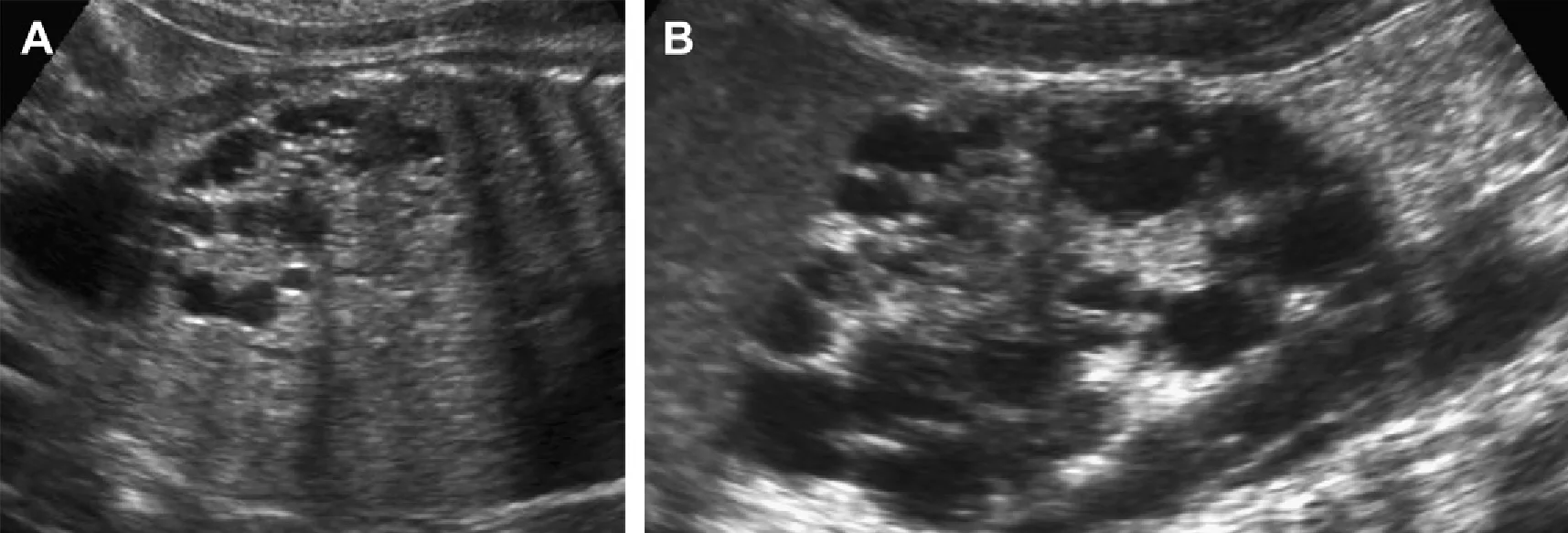
Рис. 44. Периферійні кісти при полікістозній дисплазії нирок. (А) Акушерська ультрасонограма на 33 тижні гестації показує кілька периферичних кіст в правій нирці. (В) Ультрасонограма нирок на 15-й день життя показує, що більшість кіст мають периферичну локалізацію. Ліва нирка нормальна (не відображено).
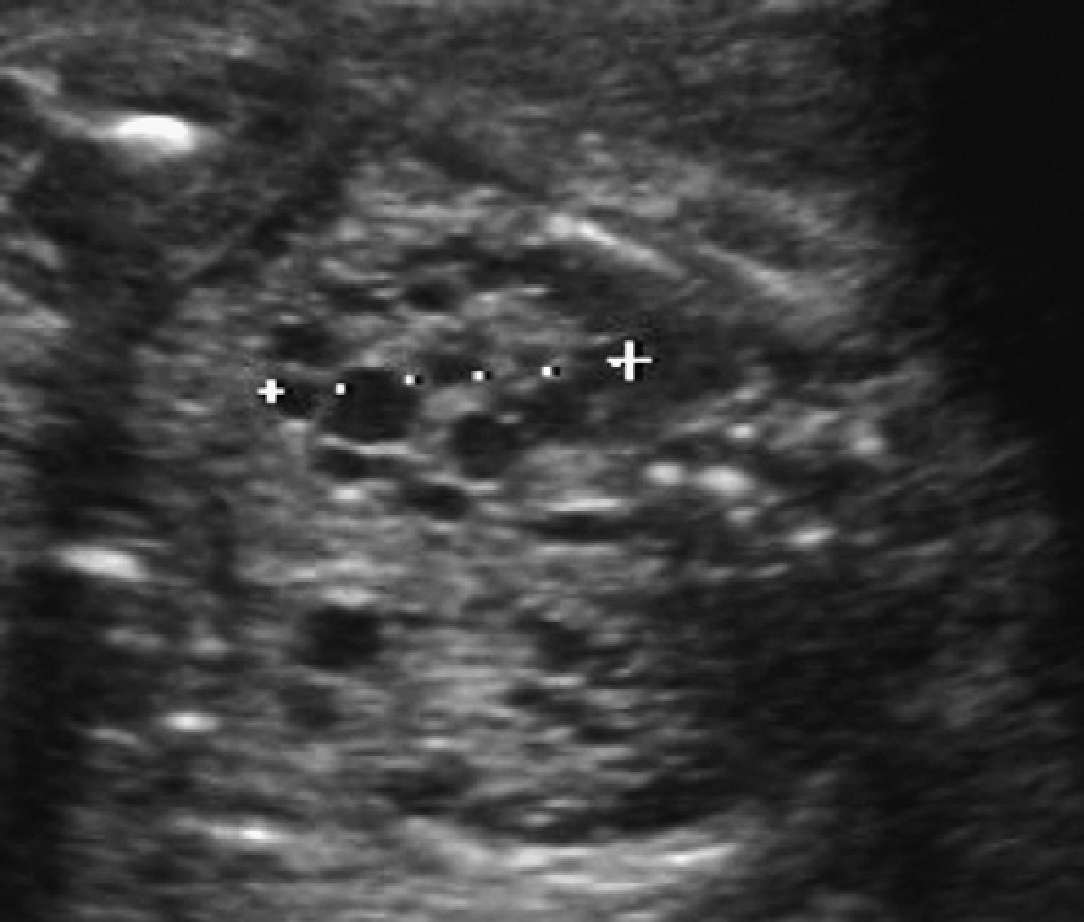
Рис. 45. Двостороння полікістозна дисплазія нирок плода (летальна форма). Акушерська ультрасонограма на 20 тижні гестації показує двосторонній периферичний полікістоз. При цьому відзначено відсутність сечового міхура і виражене маловоддя.
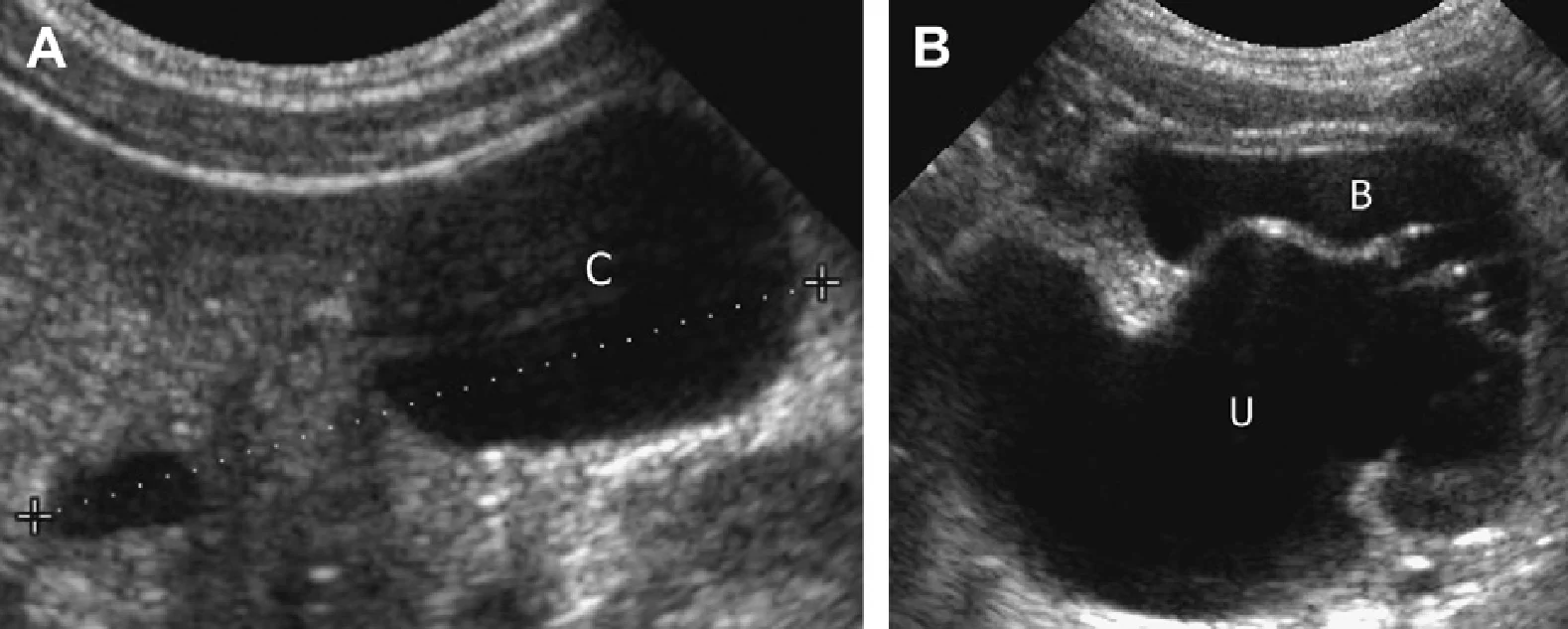
Рис. 46. Виражена дисплазія правої нирки з супутнім уретероцелє. (А) Права нирка має розміри 3,5 см в довжину і велику кісту нижнього полюса (С). (В) Іпсилатеральне уретероцелє (U) визначається в межах сечового міхура (B).
Генетика, поширеність і нозологія
Туберозний склероз є аутосомально-домінантним синдромом, пов’язаний з генами TSC1 і TSC2, які розташовані на хромосомах 9p і 16p відповідно. Туберозний склероз розвивається у 1 з 6000 живонароджених. Ниркові кісти при туберозному склерозі можуть бути пов’язані як сTSC1, так і TSC2 мутаціями, з або без супутньої делеції PKD1 гена (синдром сусідніх генів).
Туберозний склероз діагностується на основі головних і другорядних ознак. Основні ознаки включають: ангіофіброми обличчя; нігтьові фіброми; гіпопігментація, шагреневі невуси; гамартоми сітківки; ураження мозку (вузлики в корі мозку, субепендимальні вузлики, астроцитоми); кардіальні рабдоміоми; ліпоангіоміоматоз, ангіоміоліпоматоз нирок. Другорядні ознаки: ямки в зубній емалі, поліпи прямої кишки, кісткові кісти, фіброми ясен, неренальні гамартоми, кісти і карциноми нирок. Для постановки діагнозу туберозного склерозу в даний час потрібно два або більше різних типів уражень. Захворювання має тенденцію до більш серйозного розвитку у пацієнтів з мутацією TSC2.
Сонографічні особливості
Кісти нирок виникають виключно або в поєднанні з ангіоміоліпомами. Кісти мають різний розмір, кількість і розташування, охоплюють як кору, так і мозкову речовину нирки. Класичні кісти при туберозному склерозі ТНЦ можуть бути також пов’язані з гломерулокістозним малюнком нирок.
В одному з досліджень в педіатричній популяції з туберозним склерозом, виявлено 47% випадків кіст нирок, 80% випадків ангіоліпом, при цьому поєднання кісти з ангіоліпомами в 25% випадків. У більшості дітей з туберозним склерозом і нирковими кістами, при дослідженні голови виявляються ознаки, що вказують на ураження головного мозку. У деяких випадках процес обмежений тільки ураженням нирок.
Мікрокістозні захворювання нирок
Мікрокістозне захворювання нирок відносяться до вродженого нефротичного синдрому фінського типу. Поширеність вродженого нефротичного синдрому становить 1 на 10000 живонароджених в Фінляндії, і значно менше у не фінского населення.
Як аутосомно-рецесивне спадкове захворювання, мікрокістозне захворювання нирок характеризується патологічним кістозним розширенням проксимальних і дистальних канальців. При даній патології ген NPHS1 визначається на довгому плечі 19-ї хромосоми.
Сонографічно, нирки, як правило, мають нормальний розмір як внутрішньоутробно, так і при народженні, і з часом помітно збільшуються з втратою кортико-медулярної диференціації (Рис. 41).
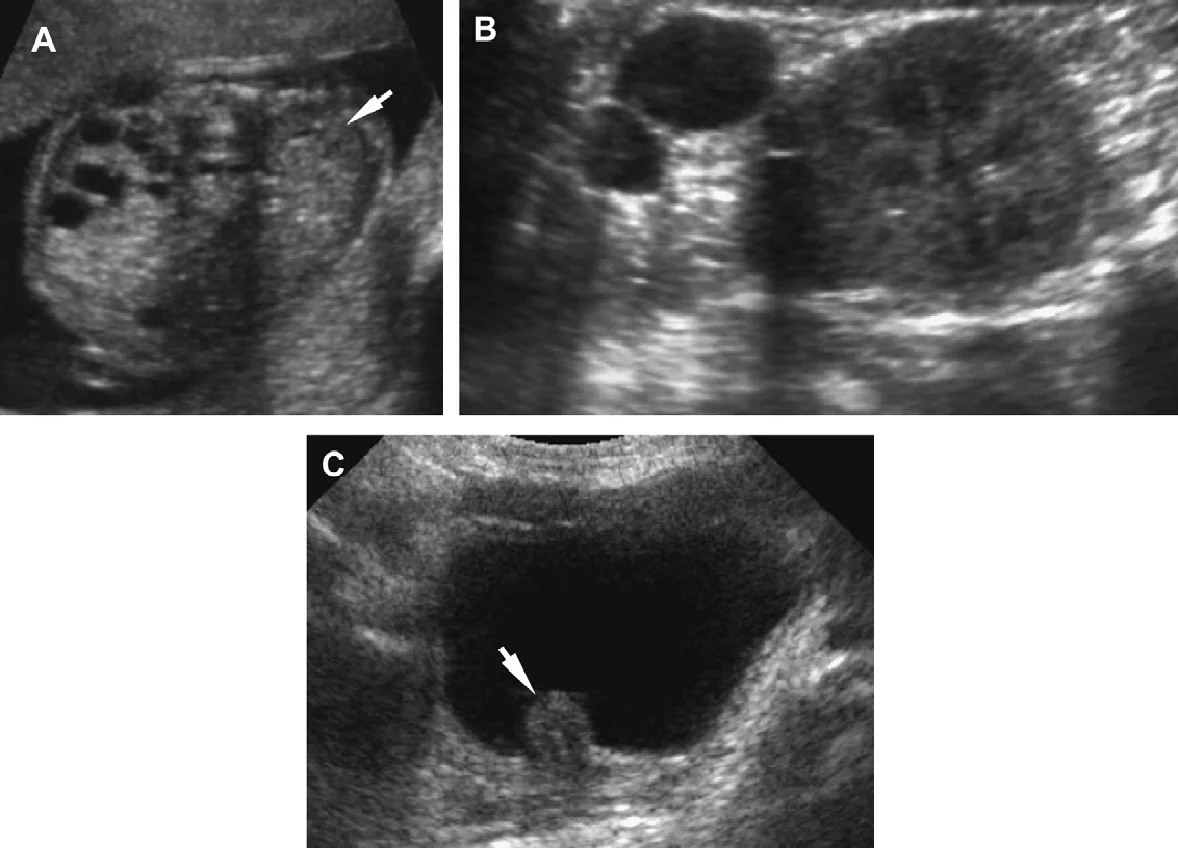
Рис. 47. Сегментарная полікістозна дисплазія нирок верхнього полюса лівої нирки. (А) Акушерська ультрасонограма на 20 тижні гестації показує полікістозну дисплазію лівої нирки. Права нирка (стрілка) – нормальна. (В) Ультрасонограма лівої нирки на 10-й день життя показує диспластичні кісти в верхньому полюсі і нормальний нижній полюс. (C) Невелике зморщене уретероцелє (стрілка) визначається в сечовому міхурі.
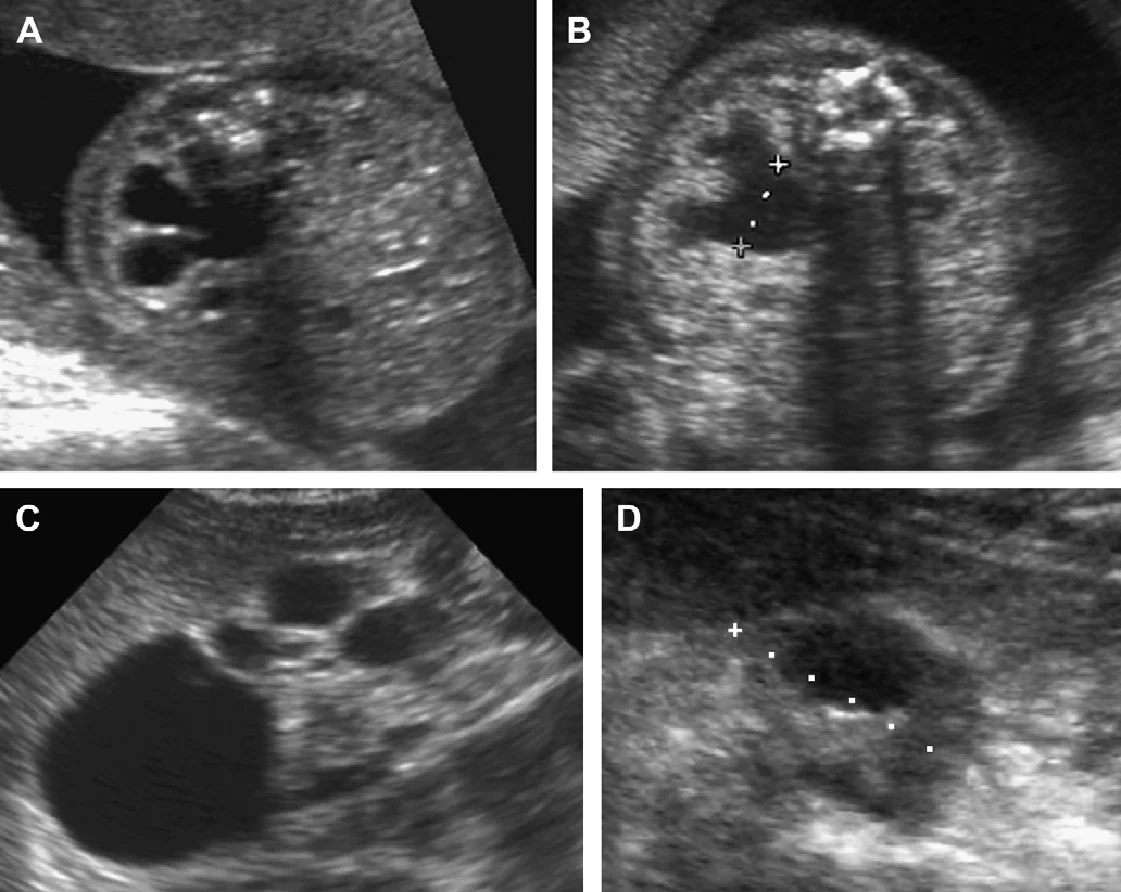
Рис. 48. Гідронефротична форма полікістозної дисплазії нирок. Акушерська ультрасонограма на 21 тижні гестації (А) і 27 тижні гестації (B) відображає ознаки можливої обструкції мисково-сечовідного сегмента правої нирки, з гіперехогенною корою нирки. (С) Ультрасонограма правої нирки в місячному віці показує характерний зразок полікістозної дисплазії нирок. Нормальна ліва нирка (не відображено). (D) У віці 1 року майже завершена інволюція диспластичної правої нирки (курсори).
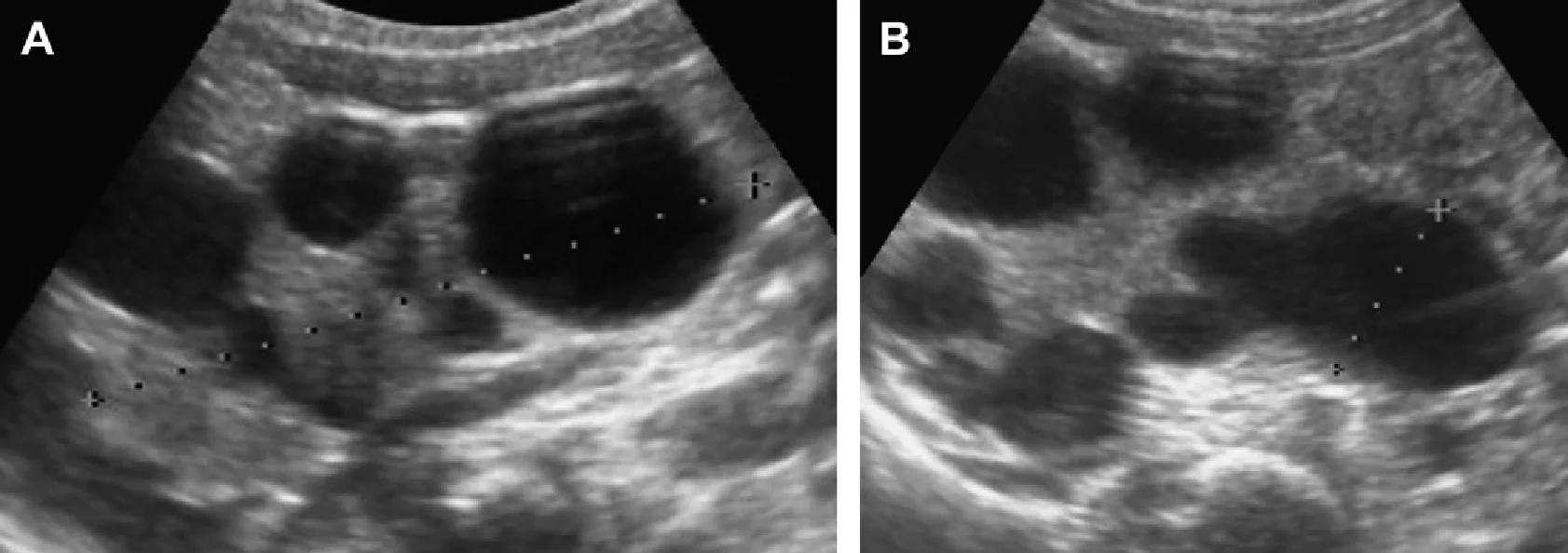
Рис. 49. Проміжний зразок полікістозної дисплазії з обструкцією мисково-сечовідного співустя. (A) Поздовжня ультрасонограма правої нирки на 2-й день життя показує типові результати полікістозної дисплазії нирок. (B) Поперечна ультрасонограма показує розширену ниркову миску (курсори) без будь-якого зв’язку з периферійними кістами.
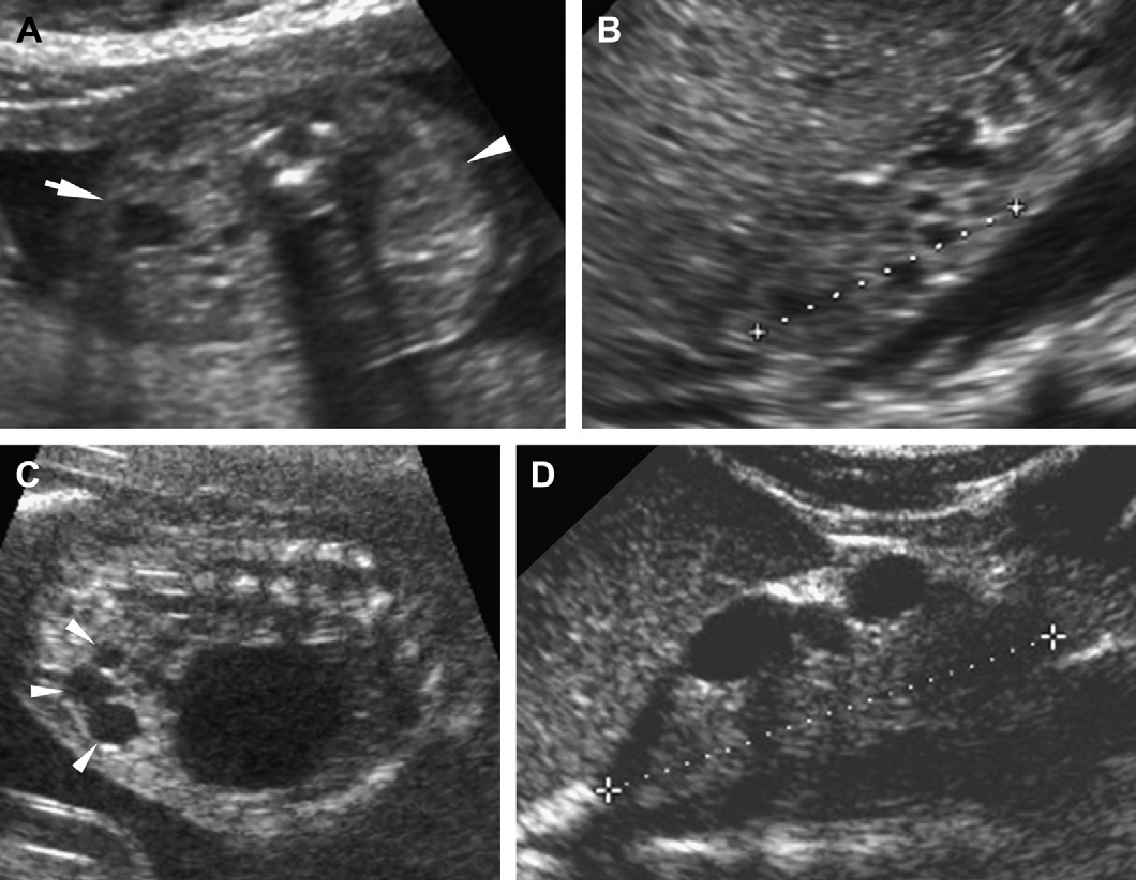
Рис. 50. Виражена дисплазія нирок у двох різних пацієнтів. Пренатальна і постнатальна картина для порівняння. (А) Акушерська ультрасонограма на 21 тижні гестації (поперечне сканування). Нормальна ліва нирка (наконечник стріли). Дисплазія правої нирки (стрілка). (В) Ультрасонограма нирки на 18 день у того ж хворого. Дисплазія правої нирки (курсори) Розміром 21 мм, нормальна ліва нирка (не відображено), розміром 47 мм. (C) Акушерська ультрасонограма на 20 тижні гестації у другого пацієнта. У правій нирці ідентифікуються три кісти (стрілки). (D) Ультрасонограма нирки в місячному віці у другого пацієнта. Виражена дисплазія правої нирки (курсори) розміром 3 см, нормальна ліва нирка (не відображено), розміром 7,1 см (компенсаторна гіпертрофія).
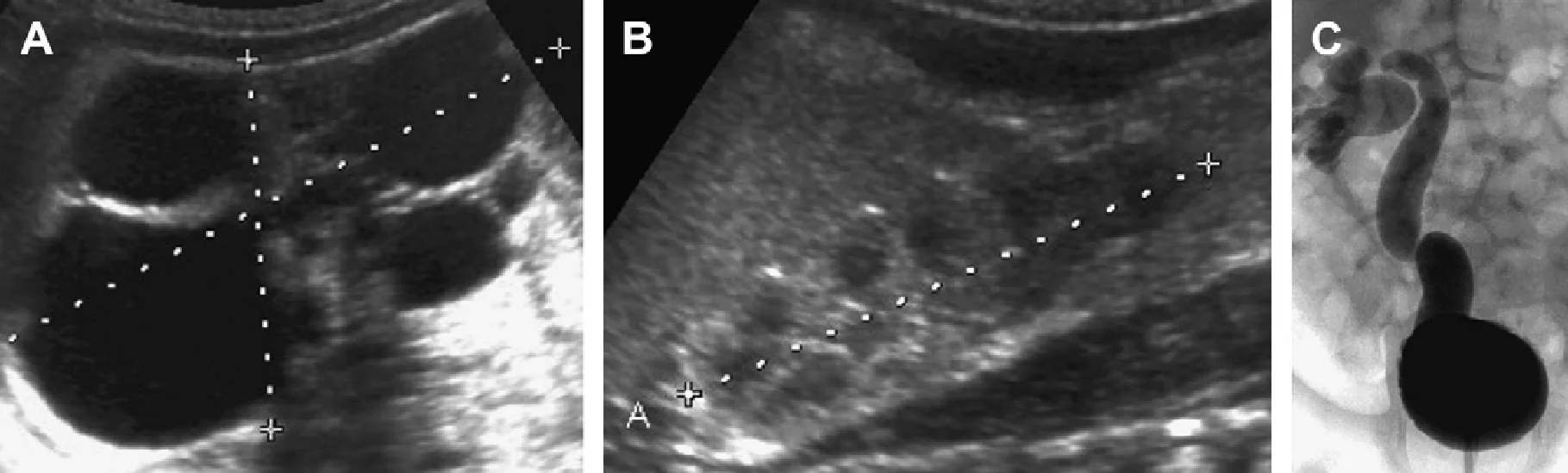
Рис. 51. Полікістозна дисплазія лівої нирки в поєднанні з правостороннім везикоуретральним рефлюксом. (А) Ультрасонограма лівої нирки на 5 день життя показує типову дисплазію нирки. (В) Поздовжня ультрасонограма правої нирки (курсори) показує невелику атрофію без дилатації збиральної системи. Виявлено переміжна дилатація дистальних відділів правого сечоводу. Цістоуретрограма (C) показує при сечовипусканні правобічний везикоуретральний рефлюкс V ступеня.
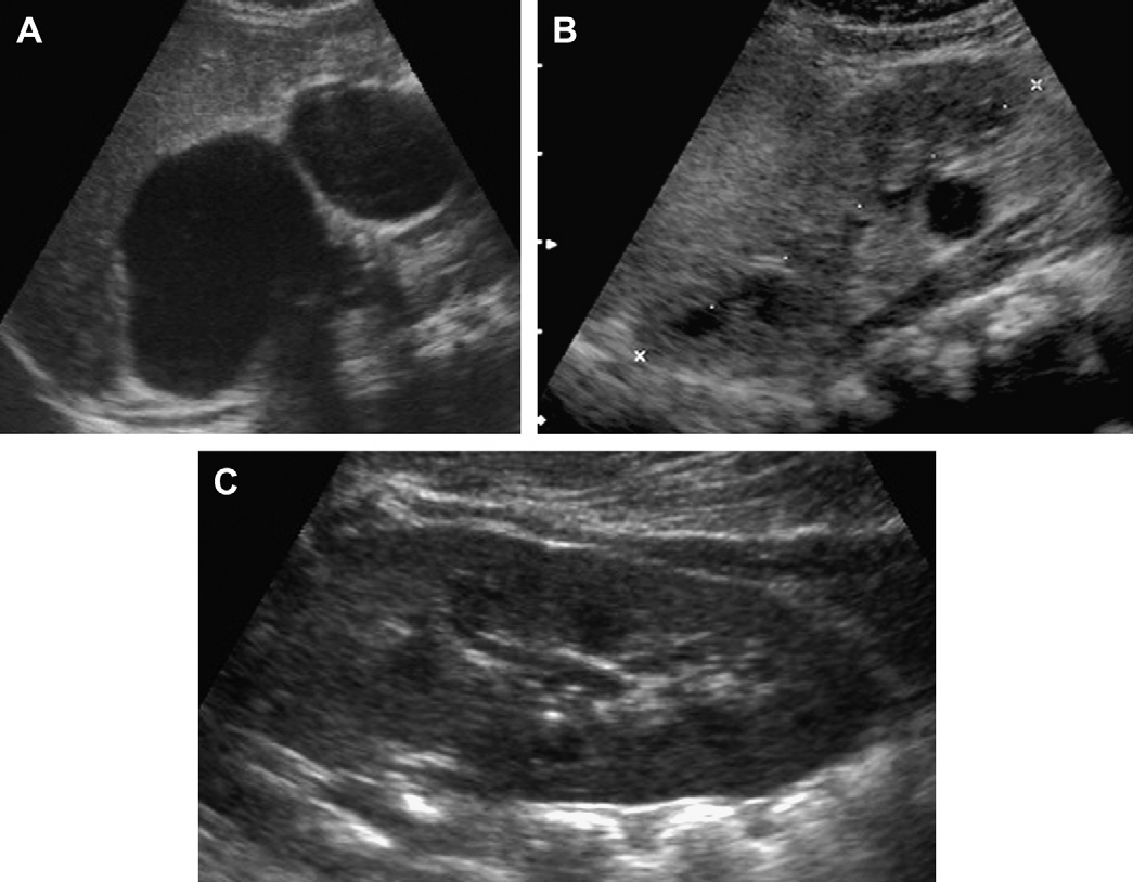
Рис. 52. Полікістозна дисплазія правої нирки в поєднанні з кістами лівої нирки. (А) Ультрасонограма правої нирки в 4-місячному віці показує дисплазію з двома макрокістами, які залишалися без змін при наступних дослідженнях (до 6 років). (B) Три ниркові кісти були зареєстровані в межах нормальної лівої нирки (курсори). (C) У віці 2-х років, ниркові кісти повністю зникли, і ліва нирка стала абсолютно нормальною.
КІСТОЗНА ДИСПЛАЗІЯ НИРОК
Патогістологія
Дисплазія нирки відноситься до аномалій розвитку нирок зі слабким диференціюванням нефронів, слабким розгалуженням і диференціюванням збірних протоків ( «примітивні канальці»), збільшенням строми, а іноді паренхіматозними кістами і метаплазією хрящової тканини.
Патофізіологія ниркової дисплазії
Нормальний розвиток результатів нирок є результатом взаємної індукції метанефричної бластеми і ампули зачатка сечоводу. Диференціація нефрона починається на 7 тижні гестаційного віку, зачаток сечоводу розширюється і ділиться дихотомічно з утворенням ниркової миски, воронки, чашок і збірних канальців. Аберантні індуктивні взаємодії між епітеліальними клітинами зачатка сечоводу і мезенхімальними клітинами вторинної нирки призводять до дисплазії. Як правило, дисплазія нирок призводить до внутрішньоутробної обструкції сечових шляхів і ампулярної дисфункції. Виразність дисплазії залежить від термінів, місцезнаходження та ступеня обструкції. Більшість кістозних дисплазій нирок є спорадичним випадком. Однак, останні досягнення в області генетики дисплазії показали, що близько 10% випадків можуть мати сімейний анамнез вад розвитку сечовивідних шляхів. При цьому виявлено причетність деяких генів до розвитку різних вроджених аномалій нирок і сечовивідних шляхів (наприклад, кодування HNF1B гена TCF2).
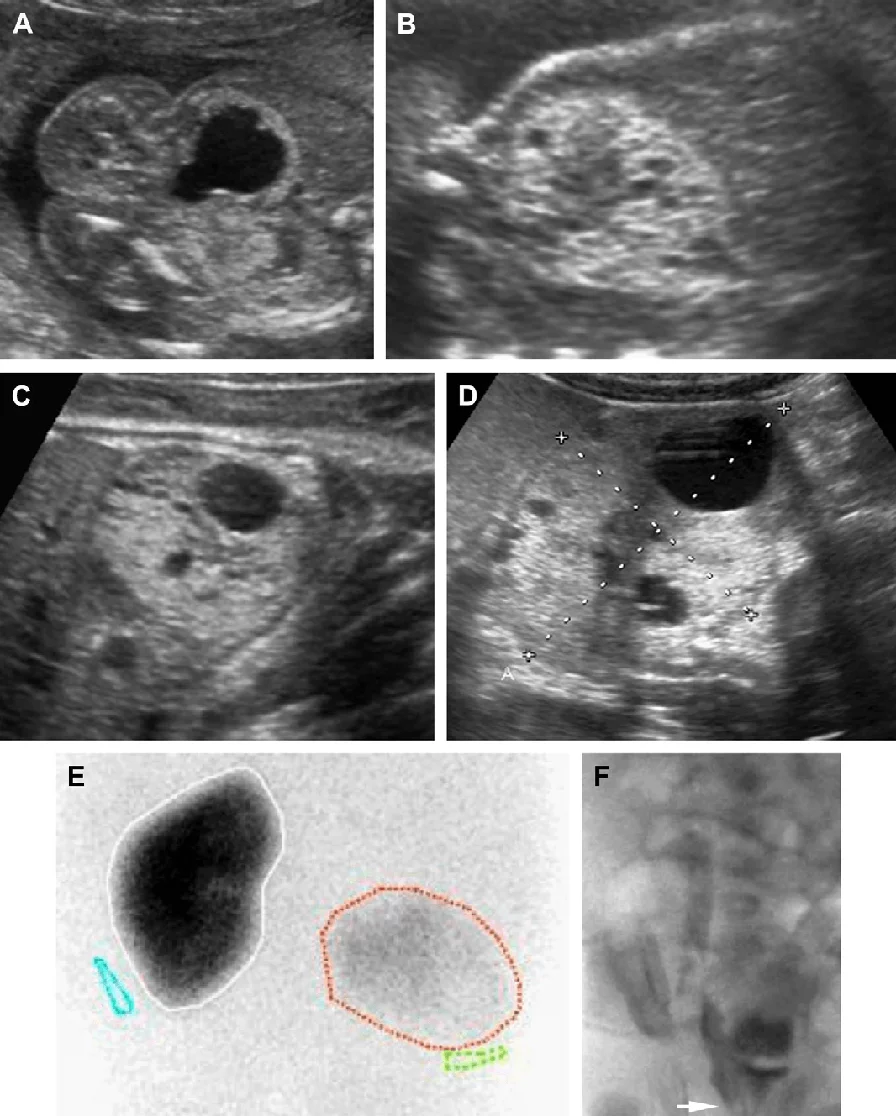
Рис. 53. Обструктивна ренальна дисплазія правої нирки, викликана ектопією мегауретера. (А) Акушерська ультрасонограма на 18 тижні гестації показує гідронефроз правої нирки з гіперехогенною ниркової корою. Ліва нирка – нормальна (не відображено). (В) Акушерська ультрасонограма на 23 тижні гестації показує невеликі паренхіматозні кісти, які тепер видно на тлі гіперехогенної правої нирки. (C) Акушерська ультрасонограма на 31 тижні гестації показує прогресування кістозної дисплазії. (D) Ультрасонограма правої нирки на 14-й день показує збільшену гіперехогенну праву нирку з макрокістозом. Ліва нирка – нормальна (не відображено). (Е) Сцинтиграфія показує практично відсутність функції правої нирки. (F) Везикоуретрограма показує мегауретер справа, який впадає ектопічно (стрілка) в піхву.
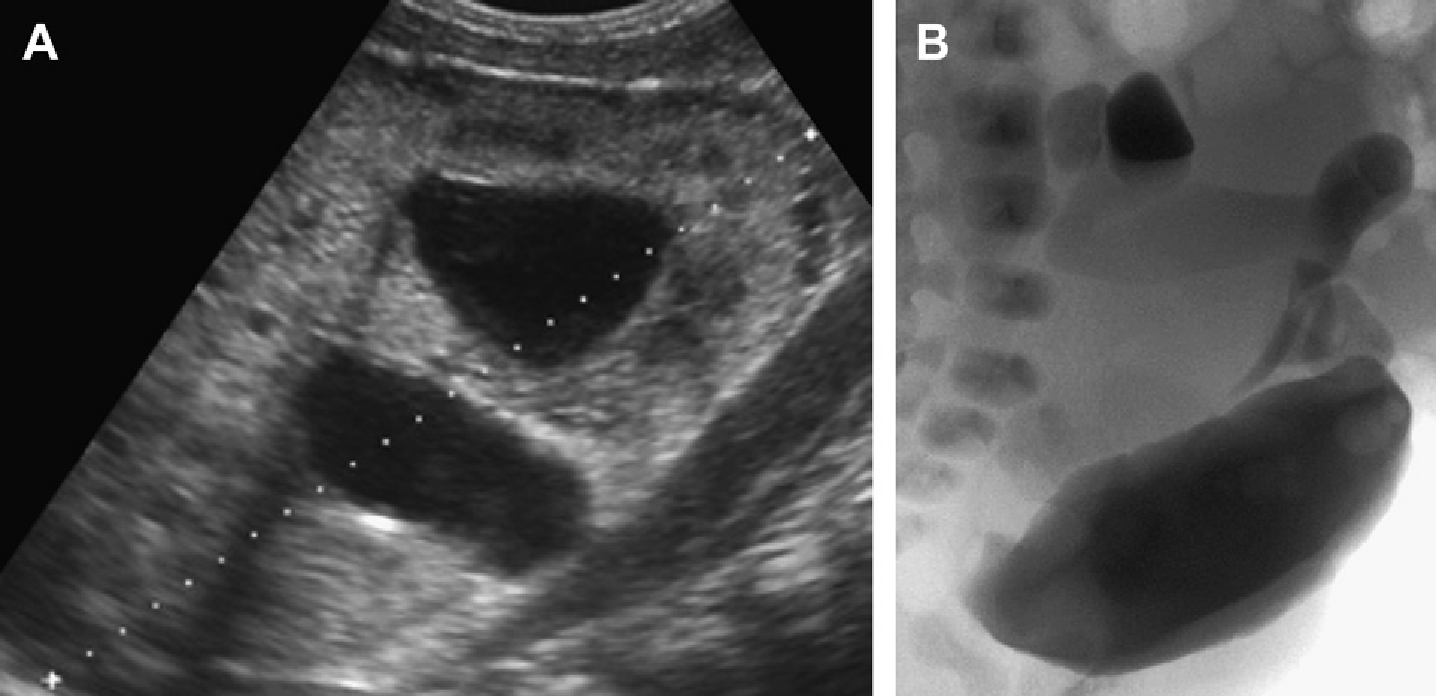
Рис. 54. Подвоєння правої нирки і мегауретер пренатально внутрішньоутробно. (А) Ультрасонограма на 9-й день життя показує подвоєння правої нирки з мегауретером (не показано) в його верхній частині і периферичні кортикальні кісти в дифузно гіперехогенній нирці. Ліва нирка – нормальна (не відображено). (B) Везикоуретрограма показує нижній полюс везикоуретрального рефлюксу.
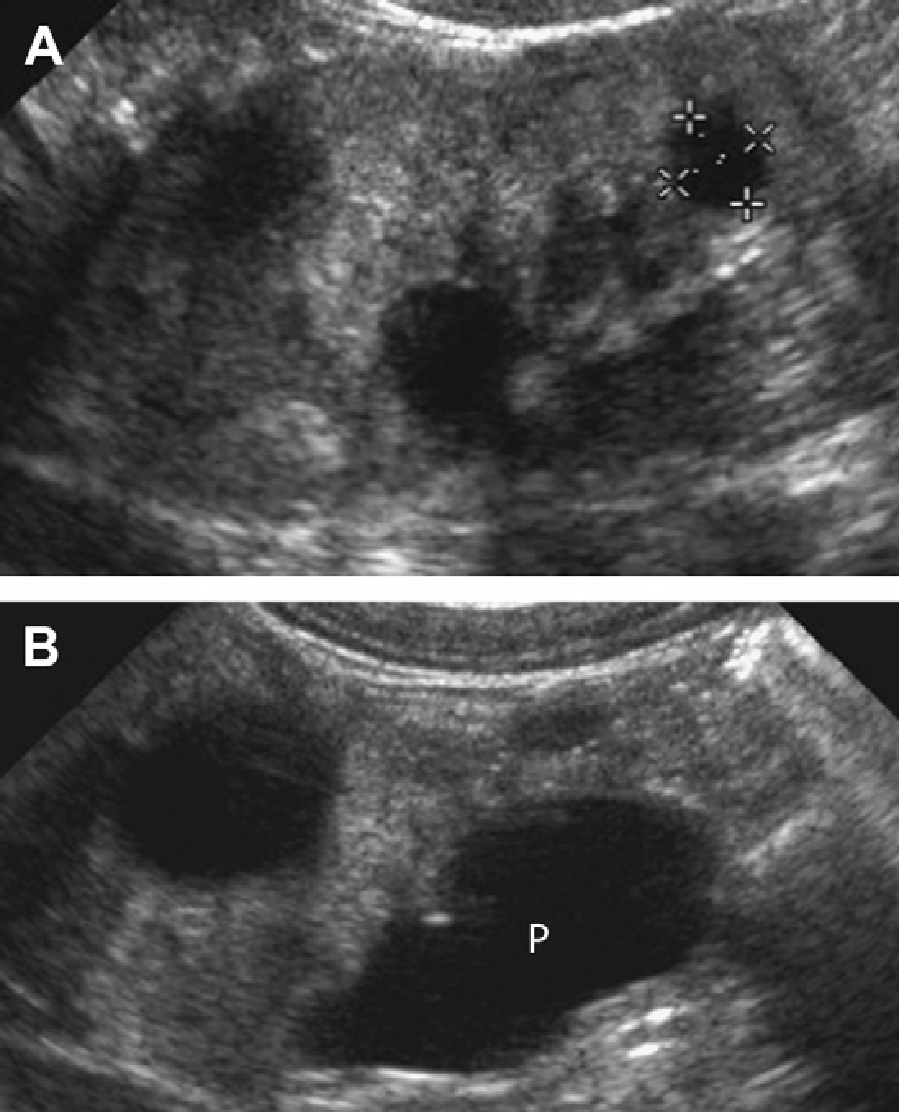
Рис. 55. Обструктивна ниркова дисплазія. 29-денне немовля з лівою ниркою, що пальпується. (A) Поздовжня сонограмма показує дисплазію лівої нирки з периферійними кістами (курсори). (B) Медіальне зображення показує дилатацію ниркової миски (Р). Сцинтіграфія не виявила наявність функції лівої нирки. Ультразвукова картина наводить на думку на гідронефротичну форму мультикістозної дисплазії нирок.
Полікістозна дисплазія нирок
Полікістозна дисплазія нирок є найчастішою аномалією черевної порожнини у новонароджених після гідронефрозу (Рис. 42-52). В даний час, полікістозна дисплазія нирок найбільш часто діагностується внутрішньоутробно (від 16 тижнів гестаційного віку до третього триместру). Через тенденцію полікістозної дисплазії нирок до інволюції внутрішньоутробно і в післяпологовому періоді, поширеність односторонньої полікістозної дисплазії нирок вище у плода (1 на 1000 – 1 на 2000), ніж у новонароджених (1 на 4000).
Двостороння полікістозна дисплазія нирок призводить до маловоддя і хвороби Поттера, і є летальною. 30% існує ймовірність розвитку контралатеральної аномалії нирок (обструкція мисково-сечовідного співустя, везикоуретральний рефлюкс, гіпоплазія) при односторонній полікістозній дисплазії.
Диференціальна діагностика полікістозної дисплазії нирок від гідронефрозу у плода та новонародженого має першорядне значення через різну тактику ведення і лікування. Сонографічно, полікістозна дисплазія нирок проявляється як кісти, які не сполучаються, різного розміру і форми всередині ехогенності ниркової паренхіми, або чітко визначаються внутрішні перетинки; великі кісти не мають передньомедіальної локалізації (тобто, відрізняються від збільшеної миски в випадках мисково-сечовивідних обструкцій); ниркові синуси не ідентифікують. Кісти випадковим чином розподілені по всій нирці, проміжна тканина не містить нормальної ниркової паренхіми. Кольорова доплерографія відображає відсутність або виражену гіпоплазію ниркових артерій. У випадках подвоєння нирки, полікістозна дисплазія нирок верхнього полюса іноді згадується як «сегментарна кістозна дисплазія».
Патогенез полікістозної дисплазії нирок був детально вивчений по серійним сонограмам у пацієнтів, які лікувалися консервативно: в 67% випадків розміри зменшилися, в 19% – залишилися без змін, в 10% відбулося збільшення розміру. Інволюція полікістозної дисплазії нирок може імітувати ниркову агенезію. Існують епізодичні дані про розвиток пухлини Вільмса та нирково-клітинну карциному на тлі полікістозної дисплазії нирок. Ця надзвичайно низька частота розвитку злоякісної патології не змінилася при консервативному лікуванні більшості дітей з полікістозною дисплазією нирок. Послідовні сонографічні дослідження показують, що у 33% пацієнтів з полікістозною дисплазією нирок відбувається повна інволюція у віці 2-х років, у 47% – в 5 років і у 59% – в 10 років.
ОБСТРУКТИВНА НИРКОВА ДИСПЛАЗІЯ
Нозологія
При обструктивній нирковій дисплазії, аномалії розвитку нирки викликано обструкцією або рефлюксом сечовивідних шляхів плода. При легкій обструктивній нирковій дисплазії, яка пов’язана з частковою дистальною обструкцією, нирки можуть бути нормального розміру зі збереженою кортико-медулярною диференціацією, або дифузно гіперехогенними з невеликими субкортикальними кістами, мегакістами і розширеними сечоводами. Тяжка обструктивна ниркова дисплазія з повною дистальною обструкцією сечовивідних шляхів (уретральна атрезія) характеризується вираженою кістозною дисплазією зі згладженою кортико-медулярною диференціацією. Сегментарна обструктивна ниркова дисплазія, яка пов’язана з подвоєною ниркою, як правило, обмежена верхнім фрагментом.
Обструктивну ниркову дисплазію також можна побачити при синдромі відвислого живота – мультисистемний патологічний комплекс чоловіків. Синдром відвислого живота включає розтягнутий живіт з надлишковою шкірою і дефектами мускулатури черевної стінки, в поєднанні з мегакістами, мегауретером, гідронефротичною дисплазією нирок і двостороннім крипторхізмом.
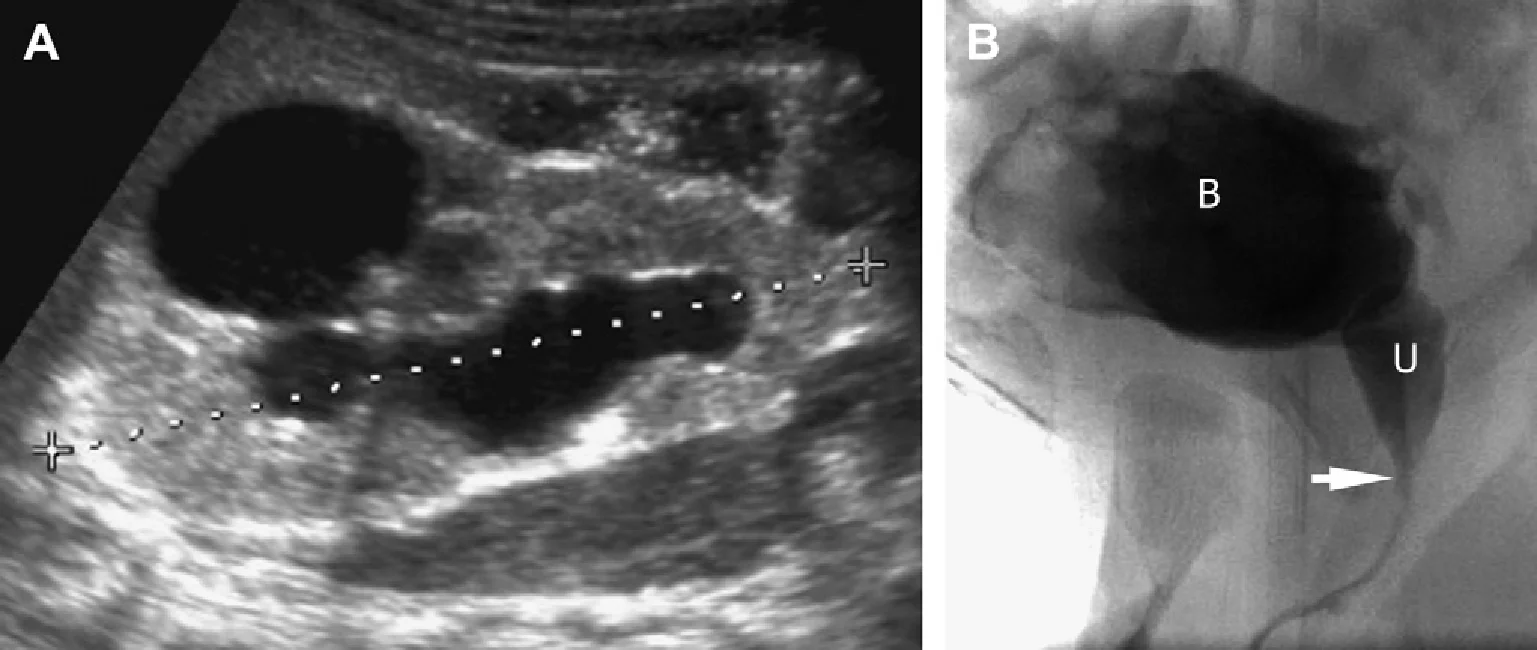
Рис. 56. Задні клапани уретри і обструктивна ниркова дисплазія. (A) Поздовжня ультрасонограма правої нирки у віці 2 тижнів показує помірний гідронефроз і велику кортикальну кісту. (В) Цистоуретерограма показує трабекулярний сечовий міхур (B) і дилатацію задньої частини уретри (U), звуження на рівні частково обтурованого клапана (стрілка).
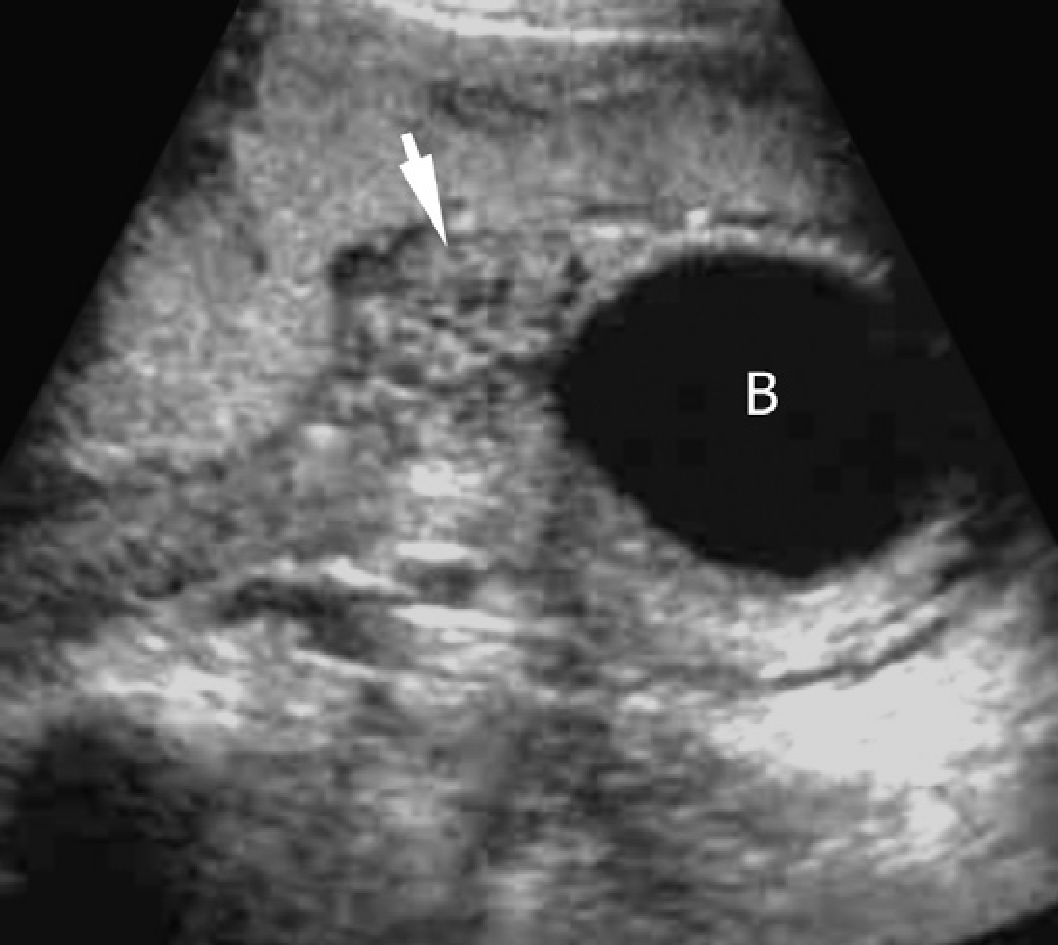
Рис. 57. Пренатальна обструктивна ниркова дисплазія за рахунок атрезії уретри. Акушерська ультрасонограма показує збільшений міхур (B) в поєднанні з вираженою дисплазією нирок (стрілка), важке маловоддя і невеликі груди.
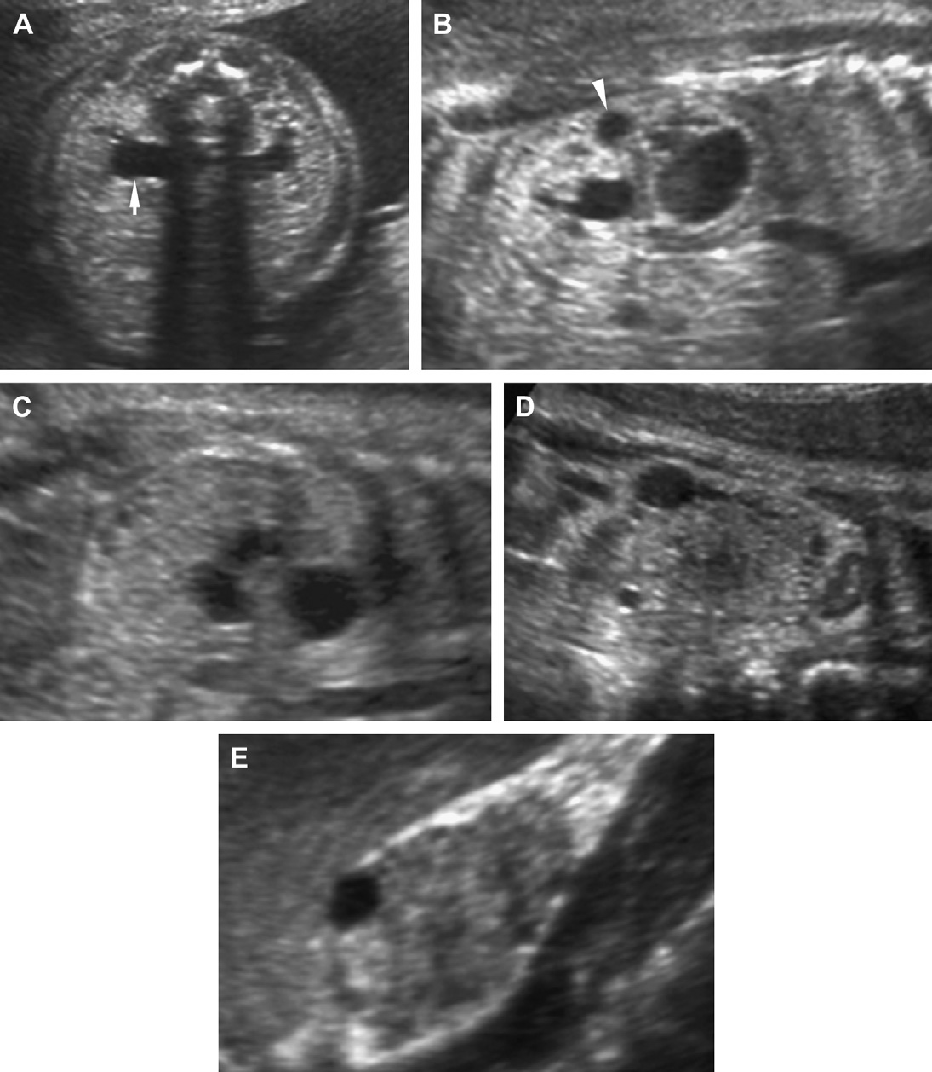
Рис. 58. Пренатально виявлена прогресивна кістозна атрофія правої нирки. (А) Акушерська ультрасонограма на 22 тижні гестації показує легку дилатацію правої миски (6 нирок, мм Стрілка). (В) Акушерська ультрасонограма на 25 тижні гестації, на якій вже візуалізуються кісти правої нирки (наконечник стріли). (C) Акушерська ультрасонограма на 29 тижні гестації показує прогресування диспластичних змін в правій нирці. (D) Акушерська ультрасонограма на 35 тижні гестації показує атрофію правої нирки. (Е) Акушерська ультрасонограма при народженні показує невелику кістозну атрофічну праву нирку. Ліва нирка нормального розміру (6 см, не показані). Везикоуретральний рефлюкс.
Сонографічні особливості
Класична антенатальная картина дисплазії нирок при сонографії включає великі яскраві нирки, з або без кістозних порожнин (вставка 4).
4 Вставка
Гіперехогенні нирки плода
– Що поєднується з дилатацією сечових шляхів: обструктивна ниркова дисплазія (з або без макрокістоза)
– Що поєднується з вираженою нефромегалією: АРПН
– Що поєднується з макрокістозом: полікістозна дисплазія нирок, синдроми, (АДПН)
– Що поєднується з характерними аномаліями: синдроми, трисомії
– Що поєднується з макросомією: синдроми надмірного зростання (Беквіта-Відемана, Перлман, Сімпсона-Голабі-Бемеля, Елеялда)
– Різне: судинні, пухлинні, метаболічні причини
– Нормальний варіант
На серійних сонограмах можна виявити затримку візуального прояву ниркових кіст всередині раніше гіперехогенної паренхіми (Рис. 53-58).
Рівномірно ехогенні нирки плода без видимих макрокіст не є рідкістю на акушерській ультрасонограмі, при цьому дуже важливо диференціювати нормальний варіант (нирки нормального розміру, помірно яскрава кора, видима кортико-мозкова диференціація після 20 тижнів гестаційного віку, нормальний сечовий міхур, нормальна кількість амніотичної рідини ) від явного захворювання нирок.
НЕГЕНЕТИЧНІ, НЕДИСПЛАСТИЧНІ КІСТИ
Прості кісти
Поширеність
Прості кісти нирок зустрічаються приблизно у 5% від усього населення будь-якої вікової групи при абдомінальному ультразвуковому дослідженні, і виникають в неураженій патологічним процесом нирці (Рис. 59-63). Захворюваність збільшується з віком: 0.22% у дітей, до 20% у 40-річних пацієнтів, і до 33% у осіб після 60 років. Подібні показники поширеності були відзначені у всіх педіатричних вікових групах.
У немовлят і дітей, прості кісти з’являються як поодинокі ураження. Кісти, виявлені внутрішньоутробно, зазвичай вирішуються до народження. Більшість простих кіст нирок у дітей залишаються незмінними при динамічному спостереженні (74%), і ведуться консервативно. Широке використання ультрасонографії в педіатрії пояснює виявлення простих кіст нирок у пацієнтів як при рутинному дослідженні при найбільш частих клінічних ситуаціях (наприклад, інфекції сечовивідних шляхів), так і при рутинному скринінгу після раніше проведеного лікування злоякісних новоутворень. Сонографічне динамічне спостереження вважається більшістю авторів необхідним для підтвердження стабільності розміру кісти і виключення рідкісних випадків АДПН, які спочатку можуть бути представлені одиночною простою кістою.
Спонгіозна нирка
Спонгіозна нирка не є генетично переданим захворюванням, характеризується ектазією сосочків збірних трубочок в мозковій речовині нирок, сконцентрованих на верхівці сосочків, за участю від однієї до всіх пірамідок. Папілярна протокова ектазія виявляється в зрілому віці при деяких вроджених аномаліях (розвитку), коли розвиваються ускладнення (утворення каменів у нирках, інфекції сечовивідних шляхів), але в більшості випадків це набутий стан (в зв’язку з цим рідкісне у дітей). Печінка при цьому має нормальну структуру. Від 13 до 19% дорослих з каменями в нирках мають ознаки спонгіозної нирки. Спонгіозна нирка є звичайним станом у дорослих, в той час як повідомлення про педіатричні випадки досить рідкісні. Двобічне ураження нирок відзначається в 70% випадків при нормальному розмірі і не порушеній функції. При дослідженні (внутрішньовенна урографія, КТ, МРТ) визначаються характерні радіальні лінійні смуги в ниркових сосочках. Ектазовані або кістозні папілярні збірні трубочки (по типу бризок з розпилювача) порівнюються з «букетом квітів». Поява папілярних смуг є ключовим діагностичним критерієм, який виявляється від 0,5% до 2% безсимптомних пацієнтів, яким проводяться клінічні дослідження з приводу різних клінічних ситуацій.
Ультразвукове дослідження в педіатричних випадках спонгіозної нирки виявляє докази легкого або помірного нефрокальцинозу в нирках нормального розміру, і нормальну архітектоніку печінки (що відрізняє її від АРПН) (Рис. 64).
Підкреслено зв’язок між спонгіозною ниркою і гемігіпертрофією: 25% пацієнтів зі спонгіозною ниркою мають гемігіпертрофію, 10% хворих з гемігіпертрофією мають спонгіозну нирку. Діти зі спонгіозною ниркою і гемігіпертрофією можуть мати неповну форму синдрому Беквіт-Відемана. Через підвищений ризик розвитку злоякісних новоутворень у пацієнтів з синдромом Беквіт-Відемана або гемігіпертрофією, ці пацієнти повинні бути періодично обстежені на предмет виявлення черевних пухлин.
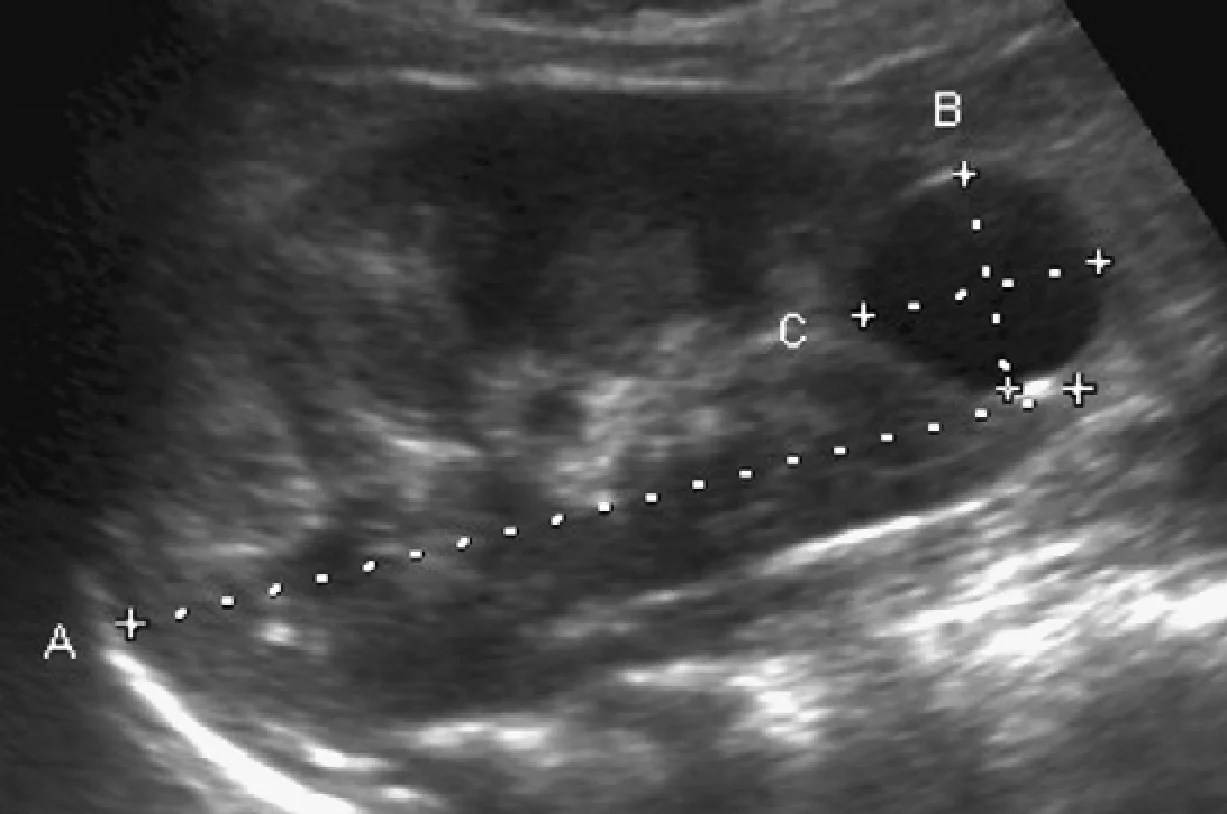
Рис. 59. Проста кіста. Дослідження однорічного пацієнта з інфекцією сечовивідних шляхів. Ультрасонограма лівої нирки показує одиночну просту кісту (курсори) в нижньому полюсі.
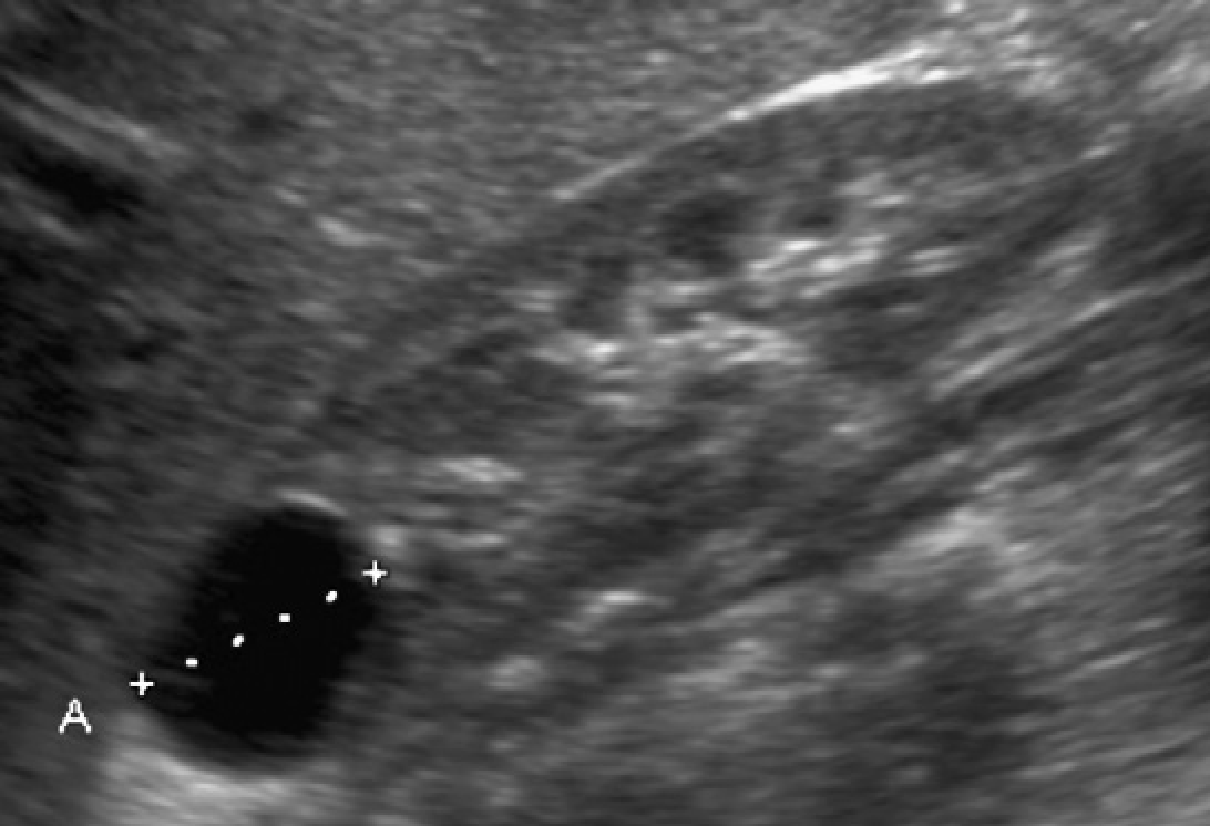
Рис. 60. Проста кіста. Дослідження дев’ятирічного пацієнта з апендицитом. Проста кіста нирки (курсори) була виявлена в верхньому полюсі правої нирки.
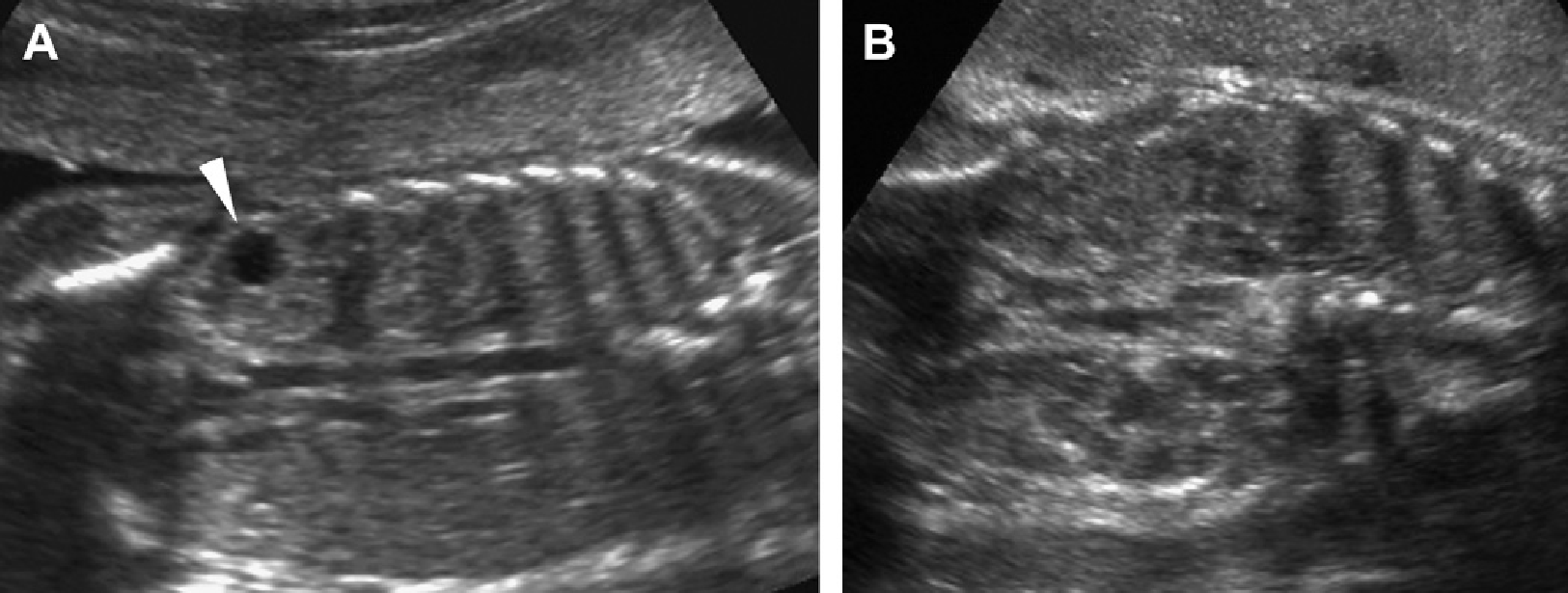
Рис. 61. Проста кіста з пренатальною елімінацією. (А) Рутинна акушерська ультрасонограма на 20 тижні гестації показує просту кісту в лівій нирці (наконечник стріли). (В) Акушерська ультрасонограма на 32-му тижні гестації показує елімінацію кісти лівої нирки. Післяпологова ультрасонограма нирок (не відображено) також була без патологічних змін.
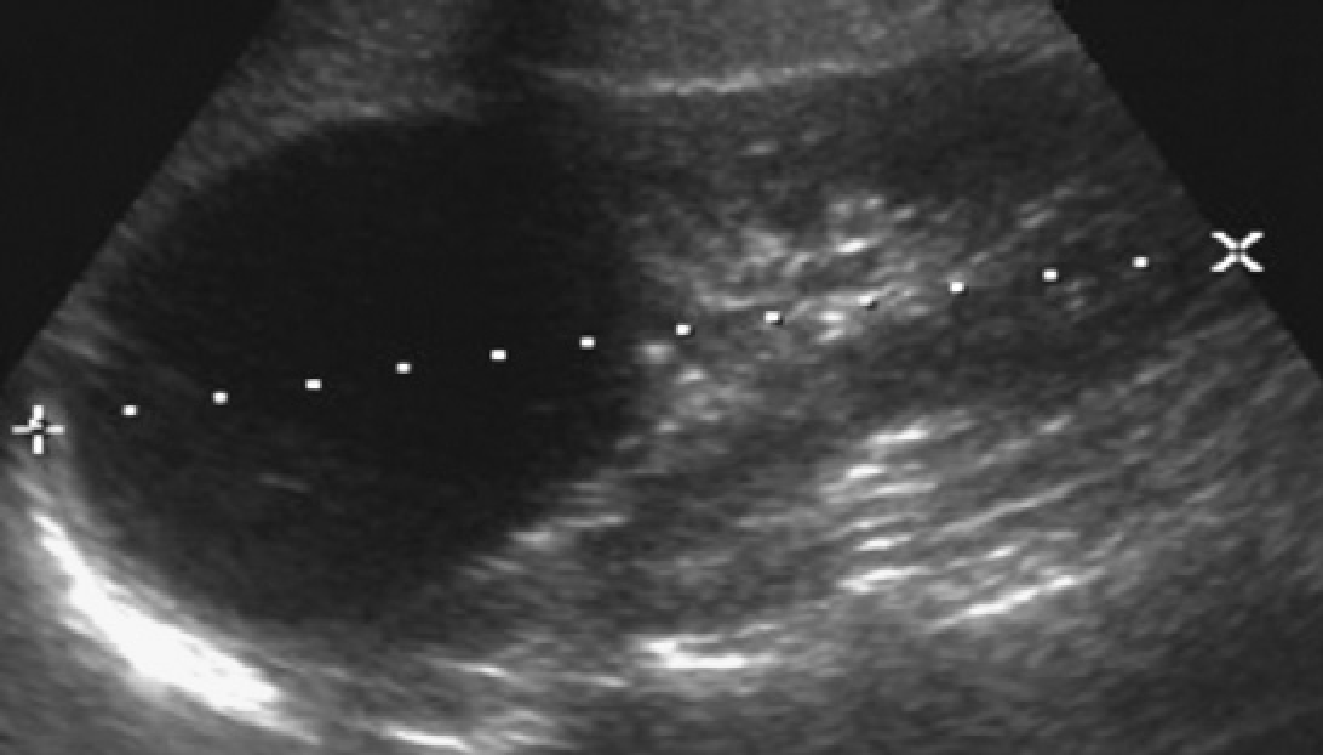
Рис. 62. Проста кіста. 14-річна дівчинка з больовим синдромом в лівому фланку. Ультрасонограма показує велику кісту в верхній частині лівої нирки. Була виконана склеротерапія in situ.
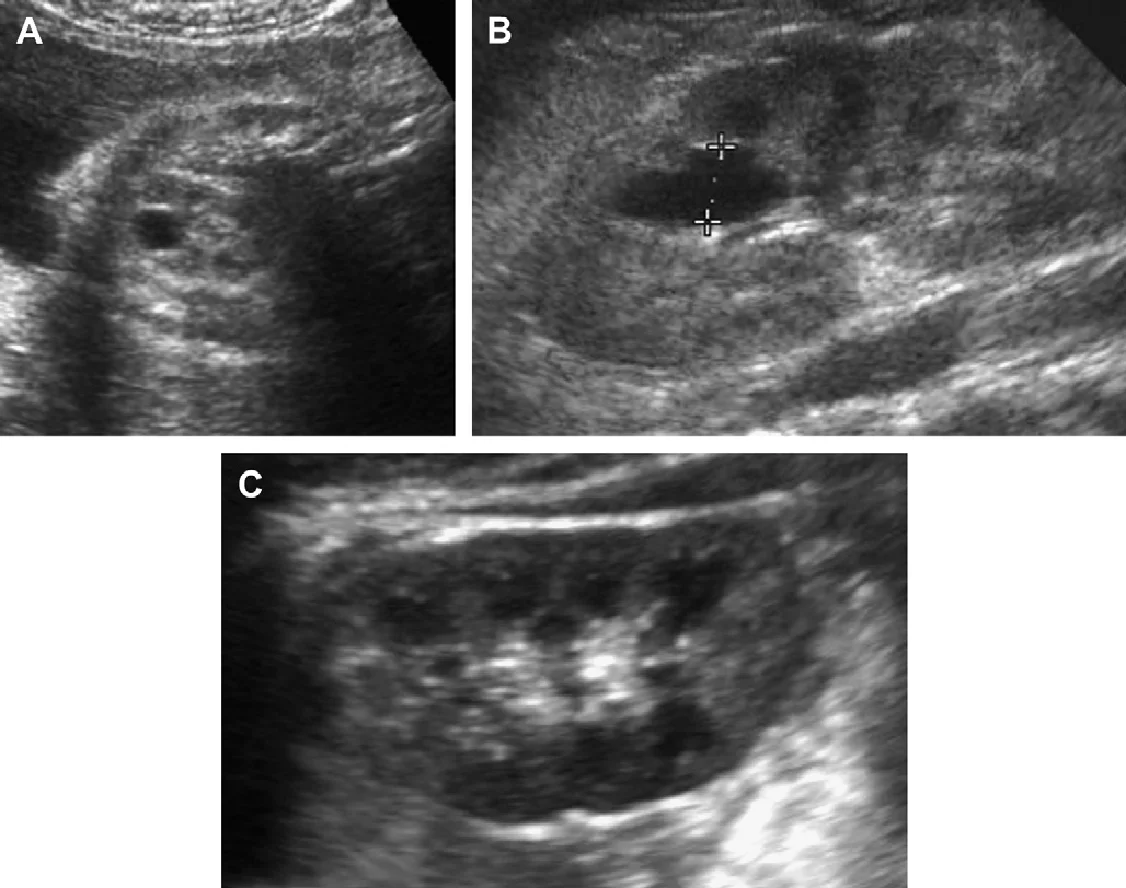
Рис. 63. Кіста ниркової чашечки або проста кіста? (А) Акушерська ультрасонограма на 32-му тижні гестації показує невелику кісту в верхньому полюсі лівої нирки. Вона залишалася без змін на сонограмі від 20 до 37 тижня гестації. (B) Ультрасонограма лівої нирки у віці 1 місяця показує кісту верхнього полюса, яка прилягає до чашечки верхнього полюса. (C) Подальша ультрасонографія у віці 1 року показує нормальну ліву нирку з елімінацією кісти.
Кістозні новоутворення нирок
Багатокамерна кістозна пухлина нирки є терміном, який відноситься до двох гістологічно різних уражень, які відрізняються в доопераційному періоді і в ході операції: Кістозна нефрома і кістозна частково диференційована нефробластома (Рис. 65-67). Кістозна нефрома (раніше «багатокамерна кістозна нефрома») є доброякісною пухлиною, яка зустрічається у дітей і у дорослих з бімодальним розподілом за віком та статтю: 65% виникають у хворих у віці менше 4 років з співвідношенням по статі – чоловічий/жіночий 2: 1 , 5% у віці від 5 до 30 років, і 30% у віці понад 30 років з співвідношенням по статі – чоловічий/жіночий 8: 1. Патологічними особливостями є: одиночне, добре оконтуроване, багатоперегородчасте утворення, з порожнинами що не сполучаються, заповненими рідиною, з внутрішньокістозними тонкими перегородками, що здавлюють ниркову паренхіму.
Кістозна частково диференційована нефробластома має такий же макроскопічний вид, що і кістозна нефрома, і такі ж ознаки при її візуалізації: при ультразвуковому дослідженні кістозна нефрома і кістозна частково диференційована нефробластома представлені як багатокамерне утворення з тонкими перегородками, без будь-яких твердих елементів. Перегородки посилюються на КТ з контрастуванням без ознак наявності твердих вузликів.
Кістозна нефрома і кістозна частково диференційована нефробластома лікуються шляхом нефректомії. При кістозній нефромі, перегородки не містять бластних клітин, тоді як кістозна частково диференційована нефробластома має ознаки септальної бластеми під час гістологічного дослідження. Кістозна частково диференційована нефробластома головним чином вражає хлопчиків і дівчаток у віці менше 2 років. Кістозна нефрома і кістозна частково диференційована нефробластома можуть бути пов’язані з плевролегеневою бластомою.
Чотири інших новоутворення дитячого віку можуть також проявлятися, як багатокамерні кістозні утворення: (1) пухлина Вільмса з утворенням кісти, яка викликана крововиливом і некрозом; (2) світлоклітинні саркоми (раніше називалися «анапластичний підтип Вільмса»); (3) кістозна аденоміосаркома (клітинний підтип) і (4) кістозний рак нирки. У більшості пацієнтів, вузлові тверді елементи також присутні на ультрасонограмах і КТ в поєднанні з кістозними компонентами (комплексні новоутворення нирки).
Інтралобарні нефрогенні залишки іноді проявляються у вигляді кістозних змін. Патологічно, кістозна пухлина Вільмса, кістозна частково диференційована нефробластома і кістозна нефрома, вважаються інтралобарними нефрогенними залишками в своїй основі. Інтралобарні нефрогенні залишки пов’язані з декількома станами, які сприяють розвитку пухлини Вільмса (наприклад, анірідія, синдроми WAGR і Denys-Drash), тоді як перілобарні нефрогенні залишки зазвичай знаходяться в пухлини Вільмса та пов’язані з гемігіпертрофією і синдромом Беквіт-Видемана. Нефробластоматоз – множинні або дифузні нефрогенні залишки, проявляється наявністю кіст в поєднанні з твердими вузликами.
Нарешті, два непухлинних захворювання можуть імітувати полікістозні пухлини нирок у дітей: сегментарна полікістозна дисплазія подвоєної нирки і, в дуже рідкісних випадках, одностороння атипова форма АДПН.
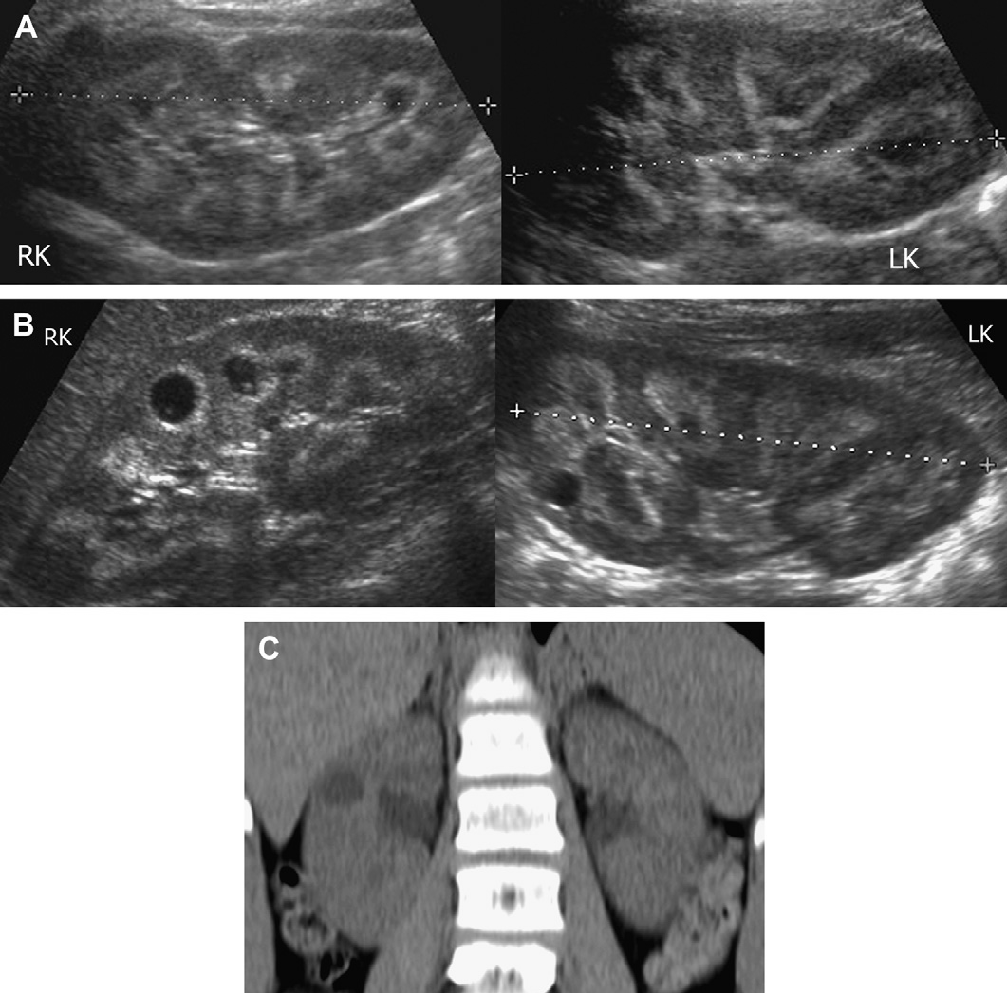
Рис. 64. Спонгіозна нирка, ймовірний діагноз, так як батьки відмовилися проводити будь-яке дослідження з контрастуванням. (А) Двостороння ультрасонограма нирок пацієнта 4 років показує дифузний мозковий нефрокальциноз уздовж периферичної частини пірамід. (В) Двобічна ультрасонограма нирок у віці 7 років показує незмінний зовнішній вигляд, за винятком двох мозкових кіст в правій нирці, і кортикальних кіст в лівій нирці. (С) КТ нирок без посилення відображає кісти нирок. Кальцифікація не визначається на КТ. Печінки – нормальна. У літературі повідомлялося про декілька випадків АДПН в поєднанні з спонгіозною ниркою.
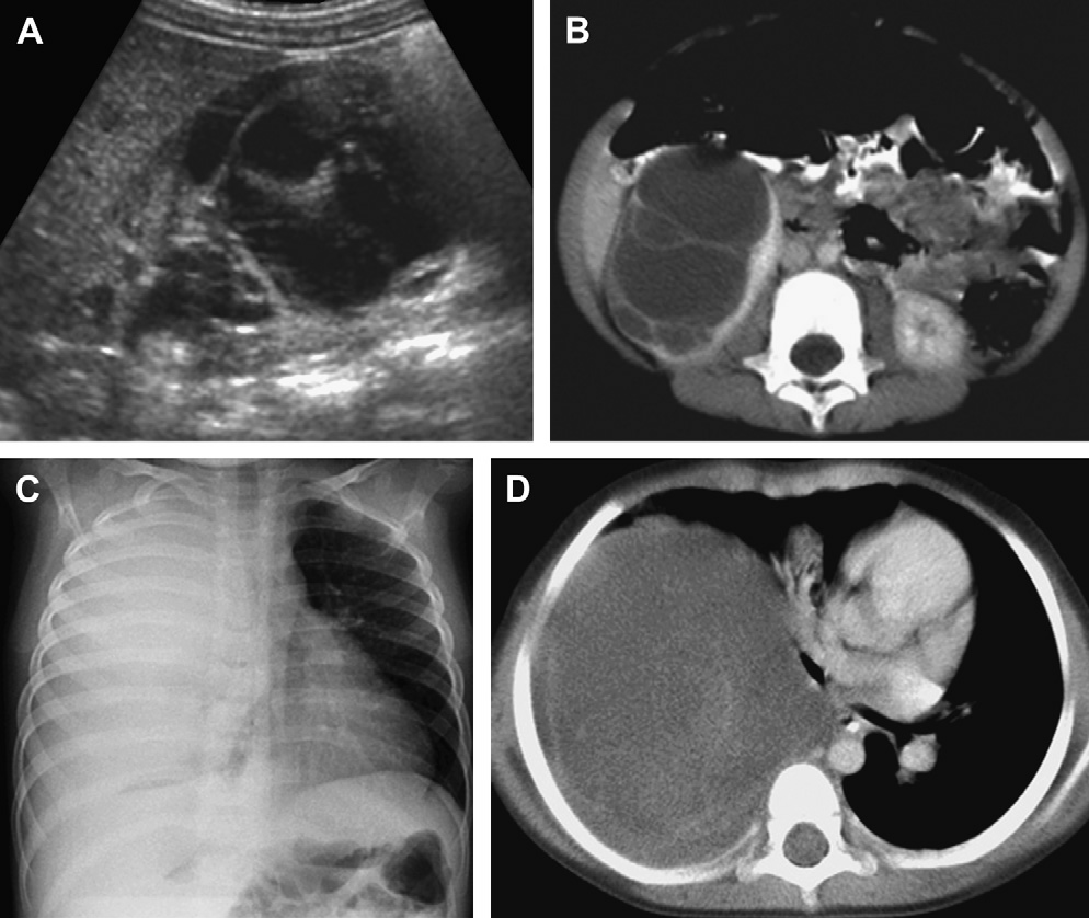
Рис. 65. Кістозна нефрома. Дворічний пацієнт з симультанною пухлиною в правій нирці і в правій легені (кістозна нефрома і плевролегенева бластома). (A) Поздовжня ультрасонограма правої нирки показує полікістозне утворення нижнього полюса. (В) КТ з контрастним посиленням не вказує на наявність твердих компонентів всередині полікістозного утворення правої нирки. (C) Рентгенограма грудної клітини показує повне затемнення правої половини грудної клітини та із зсувом середостіння вліво. (D) КТ з контрастним посиленням грудної клітини показує пухлину, яка заповнює всю праву половину грудної клітки.
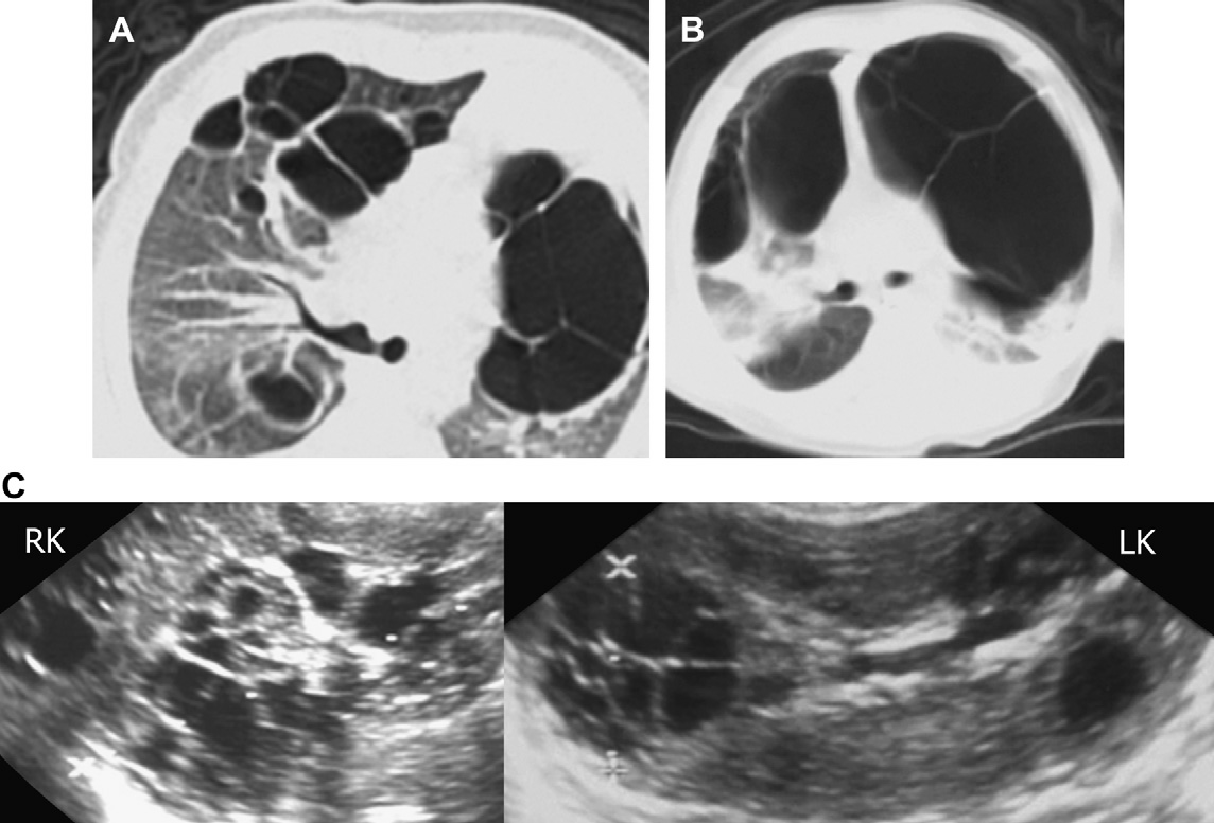
Рис. 66. Кістозна частково диференційована нефробластома і плевропульмональна бластома. Місячний хлопчик з двосторонніми легеневими кістами. Ультрасонограма нирок – нормальна. (А) КТ грудної клітини в місячному віці показує численні тонкостінні кісти. Хірургічна біопсія – неоднозначна. (В) Подальше КТ грудної клітини в 5-місячному віці показує помітне прогресування і розширення кіст. (C) Ультрасонограма нирок в 5-місячному віці показує множинні, двосторонні полікістозні утворення. Біопсія легень і нирок підтвердила двосторонню плевропульмональну бластому і двосторонню кістозну частково диференційовану нефробластому.
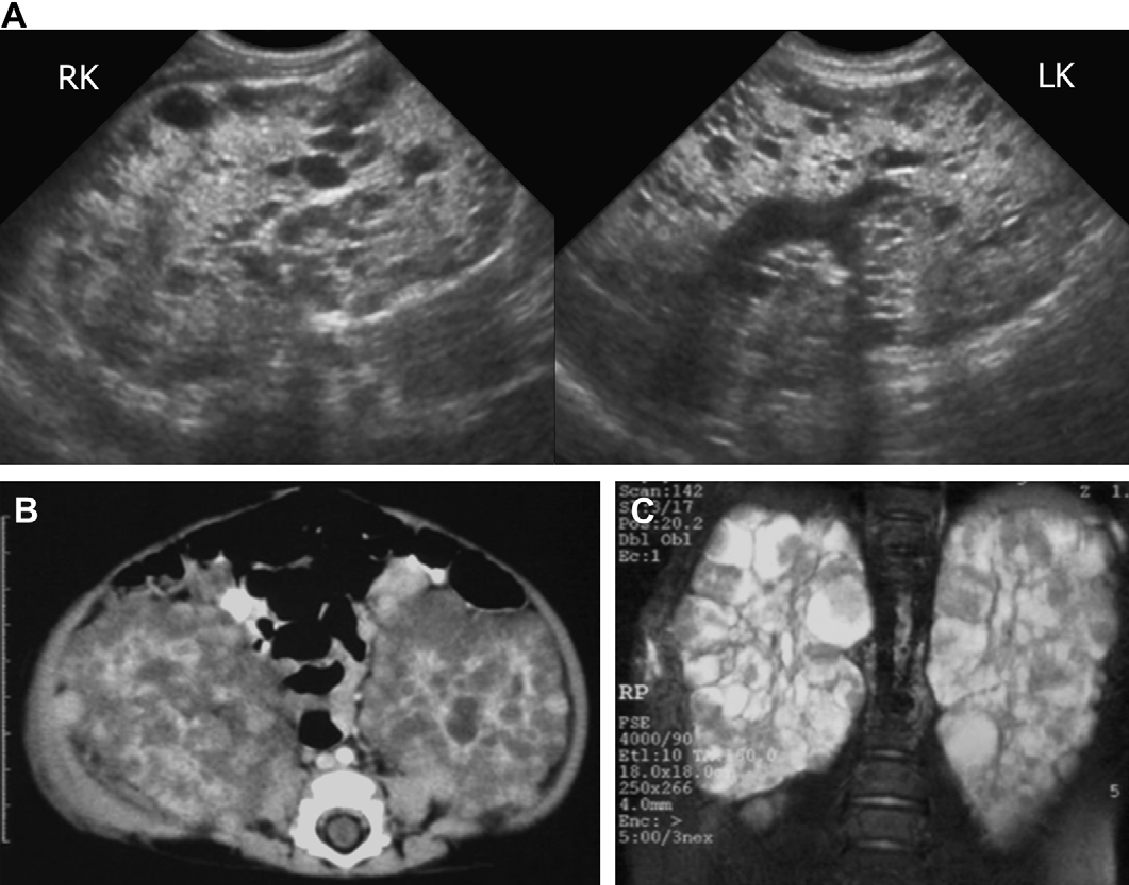
Рис. 67. Нефробластома. Трьох-місячна дитина з двосторонніми пухлинами нирок. (А) Поздовжні сонограми обох нирок показують множинні гіпоехогенні новоутворення нирок. (В) КТ з контрастним посиленням показує збільшені нирки з численними ділянками центральної і периферичної зони без посилення. (С) коронарне Т2-зважене МРТ показує дифузне залучення обох нирок з комплексом твердих і кістозних новоутворень. Хірургічна біопсія підтвердила кістозну нефробластому.
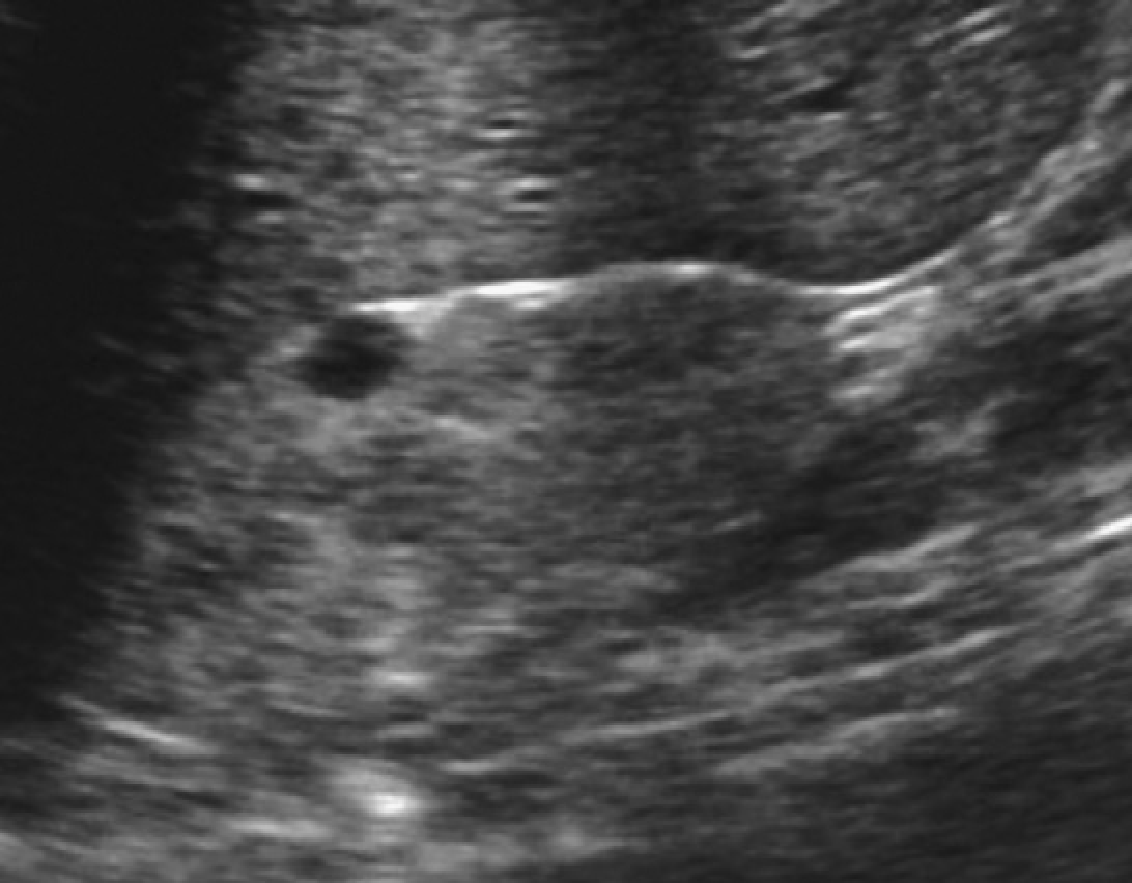
Рис. 68. Набуті кісти нирок при нирковій недостатності. Десятирічний пацієнт з термінальною стадією ниркової недостатності. Трансплантація нирки проведена через 2 роки. Поздовжня сонограма правої нирки показує невелику ехогенну нирку з набутою кістою кіркової зони.
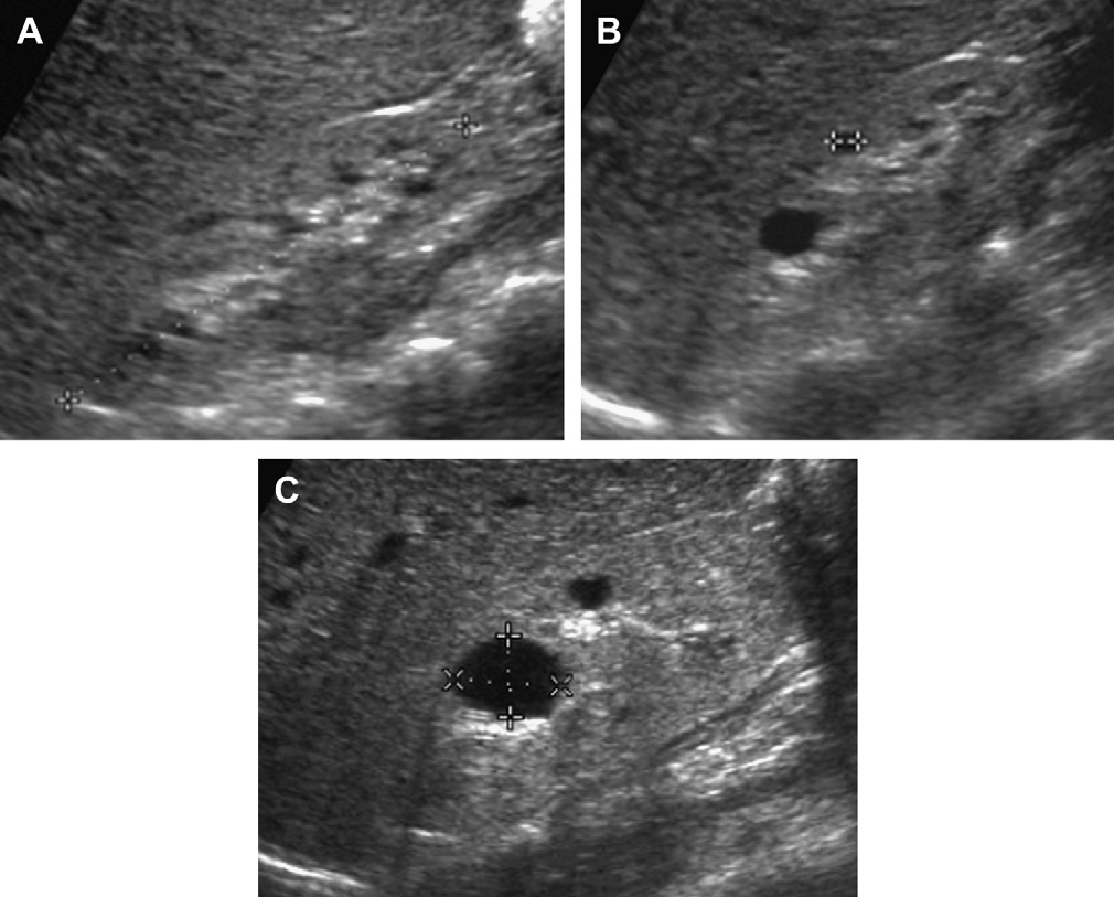
Рис. 69. Набута ниркова кіста. Пацієнт з анамнезом екстенсивної нейробластоми лівої нирки в дитинстві і гемолітико-уремічного синдрому за участю правої нирки, що залишилася. (A) Поздовжня сонограма в 5 річному віці показує помірно ерогенну праву нирку, розміром 7,1 см. (B) Сонограми в 6 років показують невеликі набуті кісти. (C) Сонограма в 7 років показує збільшення кіст (курсори), довжина нирки 7,4 см.
Набуті кісти
Набутий полікістоз нирок характеризується двосторонніми кістозними змінами, які розподілені по всій нирковій корі і мозковій речовині у пацієнтів з термінальною стадією ниркової патології і не пов’язані зі спадковими кістозними захворюваннями нирок (Рис. 68 і 69). Кісти, як правило, невеликі за розміром. Розмір нирки варіабельний, зазвичай зменшений, але іноді нормальний або навіть збільшений.
Поширеність і тяжкість набутого нирок збільшується пропорційно тривалості азотемії. Набутий полікістоз нирок визначається у 7% – 22% пацієнтів з термінальною стадією хвороби нирок до діалізу, у 58% пацієнтів на діалізі від 2 до 4 років, у 75% – на діалізі від 4 до 8 років, і у 92% – на діалізі більше 8 років. Кісти можуть регресувати після успішної пересадки нирок. Кровотеча і неопластична трансформація (ниркова карцинома) є основним ускладненням набутого полікістозу нирок.
Набутий полікістоз нирок значно рідше зустрічається у дітей з термінальною стадією захворювання нирок, ніж при азотемічної стадії у дорослих пацієнтів. Відповідно, ниркові кісти, які виявляються в педіатричній віковій групі до діалізу, ймовірніше пов’язані з основним захворюванням (наприклад, нефрофтізісом), ніж з набутим полікістозом нирок.
Є дані про те, що у дітей, набутий полікістоз нирок може розвиватися також після трансплантації печінки.
Кісти нирок нетубулярного походження
Кісти синусів нирок (кіста воріт в межах ліпоматозу синуса, навколомискова кіста лімфатичного походження) зустрічається у дорослих і рідко у дітей. Паранефральні лімфангіоми досить рідкісна патологія і зустрічаються у пацієнтів з туберозним склерозом. Субкапсулярна і паранефральна уріноми (псевдокісти сечовивідних шляхів) є зазвичай вторинними обструктивними уропатіямі, або в наслідок аномалій розвитку (задній клапан уретри, обструкція мисково-сечовідного співустя, обструкція уретро-везикального співустя), або набутими (конкременти сечоводу, травма). Чашково-мискові кісти і дивертикули розвиваються найімовірніше в наслідок аномалій розвитку, і підрозділяються на два типи. Тип I, більш поширений, пов’язаний зазвичай з малою чашкою і розташований зазвичай в полюсі нирці (особливо верхньому). Тип II являє собою дивертикул великої чашечки або миски.
Чашково-мискові кісти виникають спорадично, зустрічаються у всіх вікових групах, і, як правило, є односторонніми. При внутрішньовенній урографії, вони виявляються в 0,5% випадків. Вони інколи мають клінічні прояви, ускладнюються сечокам’яною хворобою, інфекцією або запаленням. В останньому випадку, кіста помітно збільшується по відношенню до блокованої шийки дивертикулу. Частота формування конкрементів у дивертикулах ниркових чашок знаходиться між 10% і 40%.
Резюме
Класифікація кістозних захворювань нирок у дітей заснована на відмінностях між генетичними і негенетичними причинами. Ультрасонограма є наріжним каменем візуалізації кістозної патології нирок. Оцінка розміру нирки, асиметричність залучення в процес, ехогенність паренхіми, кортико-медулярна диференціація, характеристики кісти і супутні екстраренальні аномалії є вкрай інформативними при внутрішньоутробному дослідженні, після народження і протягом усього періоду спостереження. Навідні на постановку діагнозу характерні сонографічні ознаки визначаються особливо при АРПН, полікістозній дисплазії нирок, різних синдромах вад розвитку, нефрофтізісі, обструктивній нирковій дисплазії і багатокамерній кістозній нефромі. Для проведення досліджень рекомендуємо використовувати аппарат від компании GE Voluson E8.
Питання та відповіді (FAQ)
Що таке кістозні захворювання нирок у дітей?
- Кістозні захворювання нирок – це група вроджених або набутих станів, що призводять до утворення кіст (рідинних порожнин) в одній або обох нирках. Їх класифікація базується на генетичних та негенетичних причинах.
Які основні методи діагностики кістозних захворювань нирок?
- Ультрасонограма є основним методом візуалізації кістозної патології нирок.
Які генетичні причини кістозних захворювань нирок?
- До генетичних причин належать аутосомно-домінантний полікістоз нирок (АДПН), аутосомно-рецесивний полікістоз нирок (АРПН), медулярний полікістоз, гломерулокістозне захворювання нирок та інші синдроми вад розвитку.
Які негенетичні причини кістозних захворювань нирок?
- Негенетичні причини включають ниркову кістозну дисплазію, просту кісту, губчасту нирку, кістозні новоутворення та набуті кісти (наприклад, при хронічній нирковій недостатності).
Чи завжди кістозні захворювання нирок мають симптоми?
- У дітей багато кістозних захворювань нирок можуть протікати безсимптомно. У дорослих частіше зустрічаються біль, гіпертонія та ниркова недостатність.
Які прогнози при кістозних захворюваннях нирок?
- Прогноз залежить від типу захворювання, його тяжкості та наявності ускладнень. Деякі форми можуть мати легкий перебіг, інші – потребуватимуть тривалого лікування та моніторингу.
Чи є профілактика кістозних захворювань нирок?
- Профілактика більшості вроджених форм кістозних захворювань нирок неможлива. Для набутих форм важливо своєчасно лікувати основні захворювання.